AmericanChemicalSociety.com
Reports: ND1 49109-ND1: Alkali and Alkaline Earth Metal Complexes for Catalytic Hydroaminations
The catalyzed addition of amines to unsaturated carbon-carbon bonds, the so-called hydroamination, offers a waste-free, highly atom-efficient and green pathway to produce nitrogen-containing compounds, such as amines, enamines and imines, which are valuable and industrially important bulk chemicals, specialty chemicals and pharmaceuticals. There have been significant research efforts to develop various transition metal-based catalyst systems (early transition metals of group 3-5 including the lanthanides, actinides, and late transition metals of group 8-12) over the last two decades. This research program primarily aims to develop main group metal-based catalysts with the benefit to utilize significantly less toxic elements for this industrial relevant catalytic transformation. However, significant challenges represent the labile metal-ligand interactions of main group metals; in particular facile Schlenk-type ligand redistribution processes that can potentially thwart efforts to perform enantioselective transformations. An important initial goal was therefore to develop a catalyst system that can resist such ligand exchange processes.
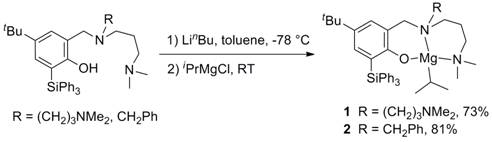
During this funding period Dr. Xiaoming Zhang has successfully investigated a series of magnesium complexes, e.g. 1 and 2, supported by potentially tri- or tetradentate triphenylsilyl-substituted phenoxyamine ligands (Scheme 1).
Scheme 1. Synthesis of
triphenylsilyl-substituted aminophenolate magnesium complexes.
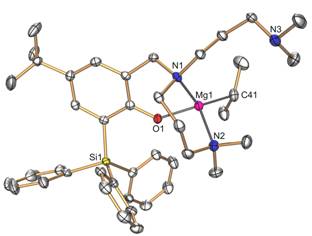
Figure 1. ORTEP diagram of the molecular structure
of magnesium complex 1. The X-ray crystallographic analysis of complex 1 confirmed a monomeric structure in which only one of the amine
sidearms is bound to the four-coordinate magnesium atom (Figure 1). The free
and coordinated sidearms in 1 undergo
an exchange process at
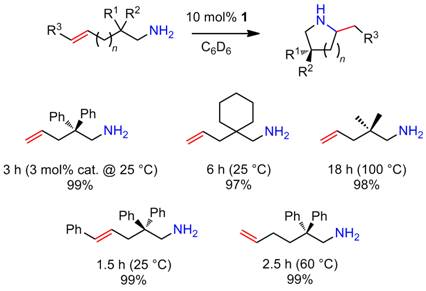
Scheme 2.
Catalytic hydroamination/cyclization of aminoalkenes using an aminophenolate
magnesium complex. Further studies are also aimed to develop catalysts based on heavier
alkaline-earth metals, e.g. calcium, which are expected to possess even higher
catalytic activity.
In a second approach to main group
metal hydroamination catalysts we are investigating the chemistry and catalytic
activity of diamidobinaphthyl dilithium complexes
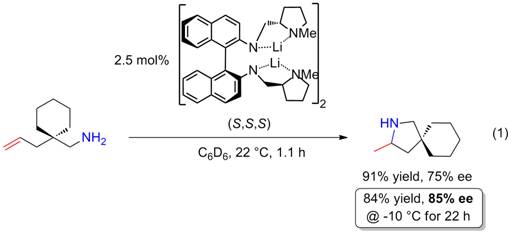
as hydroamination catalysts. Preliminary work had shown that proline-modified diamidobinaphthyl dilithium salts are viable
catalysts in the hydroamination/cyclization of aminoalkenes at ambient
temperatures with high enantiomeric excess (up to 85%; Eq. 1). These
preliminary studies suggested that the close proximity and proper geometry of
the two lithium atoms plays a pivotal role for good catalyst performance.
Catalyst systems lacking the chelating donor arms attached to the diamidobinaphthyl ligand backbone displayed significantly
diminished catalytic activity and selectivity. An important aspect of this
research program was therefore to investigate the influence of different chelating
donor arms on the behavior if this catalyst system.
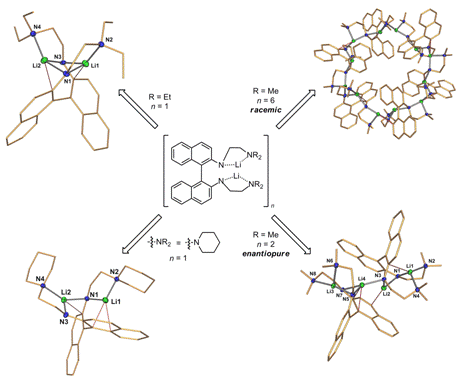
A number of novel diamidobinaphthyl dilithium complexes incorporating
various amino (Figure 2) and ether (not shown) groups in the chelating side arm
have been prepared and crystallographically characterized by Lisa Hurd. The
structures of theses species significantly depends on the steric demand of the
donor functionality. Sterically demanding piperidine or diethylamino groups
lead to monomeric diamidobinaphthyl dilithium
compounds in the solid state. A slightly reduced steric demand of a dimethylamino
group leads to a homochiral dimer in case of the enantiopure complex, while the
racemic compound forms an one-of-a-kind heterochiral cyclic hexamer .
Figure 2. Structural diversity in amino-functionalized diamidobinaphthyl dilithium complexes. Current investigations focus on the
structure and aggregation state of these species in solution and their
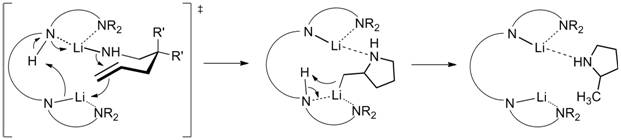
catalytic performance in asymmetric hydroamination reactions. These studies
should provide crucial information on the presumably bimetallic reaction mechanism
(Scheme 3).
Scheme 3. Proposed bimetallic mechanism for
the hydroamination/cyclization of aminoalkenes utilizing diamidobinaphthyl dilithium catalysts.