AmericanChemicalSociety.com
Reports: ND5 49221-ND5: PGM-Substituted Complex Oxide Catalysts for Selective Hydrocarbon Transformations
A variety of complex oxides synthesized by our collaborator, Prof. Ram Seshadri, with low levels of noble metals substituted on a cation site, have been studied for their reactivity in CO and methane oxidation. Since these materials are made by high temperature (typically, 700 – 1000 °C), solid-state methods, they have high crystallinity and very low surface areas. They are also thermally robust, making them interesting candidates as catalytic materials for extreme environments.
Pd-substituted BaCeO3 contains square-planar Pd(II) ions, located on the perosvkite B-sites. Each substituted Pd(II), replaces a Ce(IV), and is therefore accompanied by an adjacent oxygen vacancy. Despite its extremely low surface area (ca. 1 m2/g), the catalytic activity for CO oxidation was found to be surprisingly high. It performed better than a conventional high surface area Pd/g-Al2O3 catalyst. When O2 is present in excess, the kinetics display CO inhibition, consistent with a Langmuir-Hinshelwood mechanism in which both reactants compete for the same adsorption sites (presumably, surface Pd). When O2 is limiting, a new BaCeO3-mediated mechanism dominates the reaction. This mechanism was proposed to involve lattice oxygen available as a result of high bulk oxygen mobility in the perovskite host.
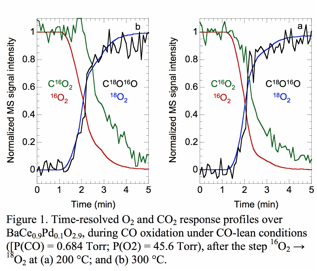
In order to probe the origin of the active oxygen, we performed steady-state isotope transient kinetic analysis (SSITKA), shown in Figure 1. An oxygen isotope step, 16O2 → 18O2, was performed during CO oxidation over Pd-substituted BaCeO3. After the isotope switch, the 16O2 signal began to decrease immediately and was 90% attenuated after 2.5 min, while the 18O2 signal increased nearly simultaneously until it attained the previous level for 16O2. There was no significant 16O18O signal, indicating that oxygen exchange between gas phase O2 and lattice O can be neglected.
Adsorbed CO can be oxidized either by oxygen adsorbed from the gas phase or lattice oxygen supplied by BaCeO3. After the isotope switch at 473 K, the C16O18O signal increased simultaneously with the 18O2 signal, demonstrating the existence of a reaction between adsorbed CO and adsorbed oxygen derived from gas-phase O2. However, we observed a pronounced time lag between the rise of the C16O18O signal and the decay of the signal for C16O2. A significant amount of 16O is therefore coming from the perovskite, rather than gas phase O2.
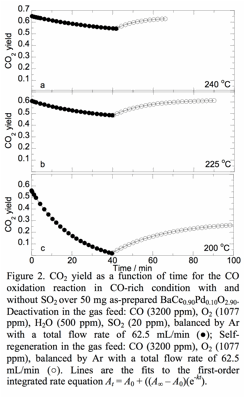
Low levels of SO2 present in the feed reversibly poison the perovskite catalyst. The extent of poisoning depends strongly on the redox stoichiometry of the feed and the reaction temperature. The effect of adding 20 ppm SO2 in the CO/O2 feed was studied under isothermal reaction conditions. Figure 2 shows that the activity recovers, either partially or completely depending on the reaction temperature, in a mixed CO/O2 gas stream when SO2 is removed. XRD and XPS studies of SO2-poisoned BaCe0.9Pd0.1O2.9 indicate that exposure to 20 ppm SO2 does not destroy the bulk structure of the perovskite. However, the surface becomes enriched in sulfates.
We have also studied La4LiAuO8 and Li2BaPdO5, two previously known oxides, as model compounds, in order to evaluate the role of isolated and immobilized Au3+ and Pd2+ ions in CO oxidation and other catalytic reactions. La2BaPdO5 appears to be an effective catalyst for CO oxidation, despite the low surface area of the oxide being used. This is the first time that a fully ordered (rather than doped) Pd2+ oxide had been used to catalyze CO oxidation. La4LiAuO8 on the other hand, is much less effective at catalyzing CO oxidation. The effect was ascribed to the low availability of lattice oxygen in this complex oxide.
Selective hydrogenation of 1,3-butadiene catalyzed by La4LiAuO8 was also studied. Both as-prepared and reduced (heated at 1000 °C in 5% H2/N2, and containing La2O3, Li2O and Au(0)) forms of the catalyst were tested, in order to compare the reactivity of Au(III) and Au(0) sites. The as-prepared material showed a low but constant conversion of 18%, whereas the activity of the reduced material declined from 73 to 32% over 5 h, due to sintering of the Au nanoparticles. The results suggest that Au(III) is not necessarily more active than Au(0) for selective hydrogenation.
This project has created opportunities for our group to collaborate with materials scientists who synthesize complex oxides, and to learn about their synthesis and characterization. It demonstrates a new paradigm in catalysis by using thermodynamically stable, low surface area materials whose activity is controlled by oxygen migration in the bulk rather than the availability of surface sites. A graduate student presented this work at the 2009 MRS fall meeting and the 2010 Gordon Research Conference on Catalysis.