AmericanChemicalSociety.com
Reports: DNI7 48917-DNI7: Reconstituting Enzymes for Direct Electron Transfer through Surface Initiated Polymerization of Conjugated Polymers
|
Figure 1. Surface-initiated Kumada transfer polycondensation. This mechanism allows for end-functionalization of the growing polymer with enzymatic cofactors. We have initially terminated polymerization with well-behaved redox probes like ferrocene in order to investigate the end-group density and electron transfer rate in these systems |
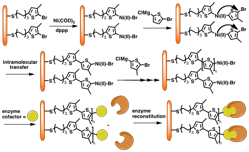
To utilize bioelectronic power sources powered through enzymatic reactions at the electrodes surface, this project is aimed at improving the electrical contact between the protein and electrode surfaces through conducting polymer wires grown through surface initiated polymerization of conjugated polymers. For most redox active proteins, the electroactive cofactors are buried deep within an insulating protein shell, which causes poor direct electron transfer (ET) between proteins and an electrode surface. The chemical nature of the linker molecule, which connects the electrode to the redox site on the protein, is important since electrons must be shuttled along some conductive pathway (or tunnel through some barrier) to the electrode surface. Self-assembled monolayers (SAMs), especially alkane thiols on gold, have been employed as linkers to attach enzymes to electrode surfaces. The electron transfer rate constant in these systems decays exponentially with distance. Enhancements in the ET rate have been observed in molecules containing delocalized bridges ("molecular wires") based on oligo(phenylene vinylene) and oligo(phenylene ethynylene) with redox active species attached to the end of the conjugated bridge. These systems have also shown distance independent ET rates up to ~ 3 nm. These molecules have also been used to interface proteins with electrodes for fundamental studies of electron transfer. The problem with conjugated tethers is that they are difficult to synthesize, have limited synthetic versatility, and become increasingly insoluble with extended conjugation length. Nonetheless, using self-assembled monolayers (SAMs) consisting of conjugated molecules, electron transfer rate constants of greater than 60,000 s-1 have been measured. This is 104 greater than measured in the best enzymatic biofuel cells to date, which rely on diffusion mediated electron transfer.
To overcome the electron transfer bottleneck in biofuel cells, the Locklin group has developed the synthetic methodology that allows for the direct growth of conjugated polymers from electrode surfaces using surface-initiated polymerization, where conjugation length (and film thickness) is controlled by reaction time and monomer concentration (Figure 1). Varying the polymerization conditions also allows for control over density of conducting polymer wires. The novel catalytic cycle in this chain growth polymerization allows for chain termination with different chemical moieties. In year two of this project, we plan to utilize these conjugated "molecular wires" to reconstitute enzymes with direct electrical contact between active site and electrode surface. These systems are important in that they can be used to study both the fundamental electron transfer kinetics in long conjugated wires and ultimately reconstitute enzymes in direct electrical contact with the electrode for efficient fuel cell operation.
The initial success in our project has involved determining conditions suitable for the formation of surface-initiated Kumada-type catalyst-transfer polycondensation (SI-KCTP). This has led to densely packed conjugated polymer films that vary in thickness from 5-50 nm. In order to completely control the polymerization methodology, there is a considerable amount of work remaining to fully understand the mechanism of polymer growth and thereby optimize the conditions for film formation. This is crucial in order to fully utilize this technique for direct electrical contact in enzymatic biofuel cells, in which control over chain orientation and surface morphology are critical at optimizing device performance.
The mechanism of KCTP in solution is still under considerable debate and may differ from that found using SI-KCTP. For example, evidence suggests that the reductive elimination step is rate-determining for KCTP in solution, but our observations suggest that the mechanism may differ with surface bound catalyst systems in SI-KCTP. Our preliminary results have shown that film growth from densely packed monolayers is strongly hindered by the steric bulk of the monomer, and that the growth can be enhanced by activation of the Grignard monomer with lithium salt. These factors lead us to speculate that transmetallation of the surface-bound catalyst center is the limiting factor for chain-growth polymerization, and the optimization of transmetallation will yield a polymerization with greatly improved control and chemical diversity. We have also observed that certain aryl-bromide based monomers that polymerize efficiently in the solution phase do not yield polymer in SI-KCTP. This fact, in light of the critical role that rapid oxidative addition plays in the catalyst-transfer step which leads to a chain-growth polymerization, suggests that oxidative addition may become rate-limiting in SI-KCTP of aryl bromides, particularly when the aromatic ring is electron rich and in the absence of heteroatoms which serve to weaken the carbon-halogen bond.
