AmericanChemicalSociety.com
Reports: DNI3 49914-DNI3: Renewable Energy Catalysis: The Study of Water Oxidation by Binuclear Metal Complexes
Summary:
Solar energy is the largest exploitable renewable energy resource and there is a large interest in developing molecular systems that can capture solar energy and use it to produce oxygen and hydrogen from water. Nature uses solar energy to catalyze the oxidation of water to O2 at a tetrametallic manganese cluster; however, the most efficient artificial water oxidation catalysts employ rare and expensive metals (e.g. Ru, Pt, Ir) and thus are not suitable for large scale solar energy conversion. We have proposed to investigate an approach for metal-assisted water oxidation based on inexpensive, first row transition metal complexes that employ a novel paradigm of O2 formation through the reductive elimination of a bis(m-oxo) dinuclear metal complex.
For the past 12 months we have been successful in synthesizing ligands based on two of our bridging linker frameworks, survey their coordination chemistry to generate Ni, Cu, and Co complexes, and investigate the electrochemistry and water oxidation reactivity of the corresponding bis(m-hydroxo) dinuclear complexes. Preliminary results suggest that such complexes may be able to oxidize water efficiently. We are currently extending our studies to other transition metal ions that can access high oxidation states and can be employed in various small molecule activation processes.
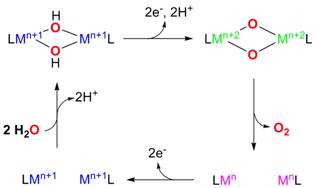
Scheme 1. Targeted electrocatalytic water-oxidation cycle using a binuclear metal complex.
While several reaction schemes have been considered for water-oxidation model systems, the formation of O2 through the reductive elimination of a bis(m-oxo) binuclear complex, to the best of our knowledge, has not been employed before for water-oxidation catalysis. We are targeting the development of dinuclear metal complexes designed to catalyze water oxidation by employing the latter reaction pathway for O2 formation (Scheme 1). Specifically, binuclear metal complexes with water-derived hydroxide bridging ligands will be oxidized electrochemically to generate the bis(m-oxo) dinuclear metal species, which will undergo reductive elimination to generate O2.
The targeted ligands were binucleating variants of the tetradentate ligand, tris[(2-pyridyl)methyl]amine (tpa); use of different rigid linkers will allow the control of the metal-metal distance by modulating the spatial distribution of the two metal centers. We were successful in synthesizing the binucleating ligand xanthene-bis(tris(2-pyridylmethyl)amine), X-(tpa)2, as well as the dibenzofuran-bridged equivalent (Figure 1, boxed ligands). Moreover, the developed synthetic route gave us access to ligand derivatives that employ 6-methyl-pyridyl groups in 30% to 50% yields for a six-step sythesis. For each ligand framework we have synthesized six ligand variants with one, two, and three methyl groups in the 6-pyridyl position (R1, R2, R3 = H or CH3, Figure 2), which will allow us to further control the reactivity of the targeted metal/O2 intermediates.
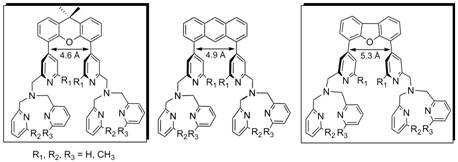
Figure 1. Proposed ligands for dinuclear metal complexes (the boxed ligands were successfully synthesized).
The ability of the synthesized ligand X-(tpa)2 to form dinuclear complexes was tested initially in reactions with Cu and Ni salts. Interestingly, when Bu4NOH was added to a MeOH solution of X-(tpa)2 and Cu(ClO4)2, a (m-hydroxo)dicopper(II) complex was formed (Figure 2). The crystal structure confirms the ability of our ligand to support dinuclear complexes with water-derived bridging ligands. By contrast, addition of Ni(ClO4)2 and Bu4NOH to a MeOH solution of the ligand generated a bis(m-hydroxo)dinickel(II) complex (Figure 2). While the cyclic voltammetry (CV) experiments reveal that the oxidation of the [(X-(tpa)2)Cu2(OH)](ClO4)3 complex occurs only at quite high potential (~1.2 V vs. Fc/Fc+), the CV trace of the [(X-(tpa)2)Ni2(OH)2](ClO4)2 complex shows two oxidation waves at 0.47 V and 0.97 V vs. Fc/Fc+ that have been assigned to the Ni2(II,II)/(II,III) and Ni2(II,III)/(III,III) redox couples, respectively (Figure 3).
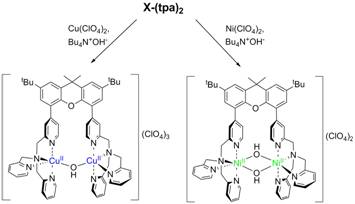
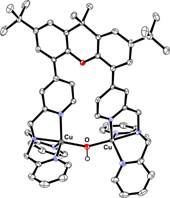
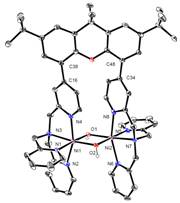
Figure 2. Synthesis and crystal structures of dinuclear Cu and Ni hydroxide complexes with the X-(tpa)2 ligand.
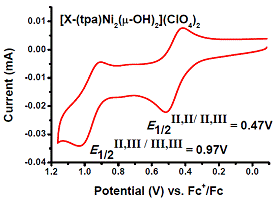
Figure 3. Cyclic voltammetry of [(X-(tpa)2)Ni2(OH)2](ClO4)2 in MeCN (100 mV/s scan rate).
The [(X-(tpa)2)Ni2(OH)2](ClO4)2 complex exhibits an interesting reactivity in air when is quickly changing its color from blue to purple. X-ray characterization of the purple product reveals the formation of the carbonate-bridged complex [X-(tpa)Ni2(m-CO3)(H2O)](ClO4)2. Interestingly, this CO2 binding process is reversible in presence of aqueous hydroxide (Figure 4) and we are currently investigating its CO2 fixation potential.
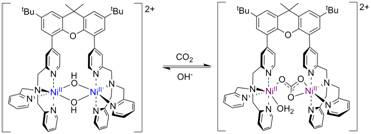
Figure 4. Reversible binding of CO2 by [(X-(tpa)2)Ni2(OH)2](ClO4)2.
Overall, these initial results confirm our assumption that bis(m-hydroxo) bimetallic complexes have accessible oxidation potentials and under the appropriate conditions should allow the formation of high-valent complexes. In the case of Ni complexes, it seems that the formation of a Ni(III)Ni(III) intermediate can be achieved, which will be essential for achieving water oxidation. Interestingly, preliminary results show that the [(X-(tpa)2)Ni2(OH)2](ClO4)2 in presence of strong chemical oxidants can lead to formation of up to 50% of O2, as measured by a fluorescent sensor. We are currently investigating the electrochemical oxidation of various Ni complexes in an attempt to oxidize water catalytically.
While designing various ligands that can stabilize transition metal ions in higher oxidation states, we have found that the tetradentate ligand N,N'-ditertbutyl-2,11-diaza[3.3](2,6)pyridinophane, N4, is exhibiting an interesting reactivity behavior. As part of this work, we have recently reported (J. Am. Chem. Soc. 2010, 132, 7303-7305) that N4 stabilizes Pd(III) centers and allows for the isolation and characterization of the first mononuclear organometallic Pd(III) complexes [(N4)PdIIIMeCl]+, [(N4)PdIIIPhCl]+, and [(N4)PdIIIMe2]+. These complexes undergo a light-induced elimination of the homocoupled products ethane or biphenyl, the observed formation of ethane from monomethyl Pd complexes being unprecedented (Figure 5). In light of other recent studies that have proposed Pd(III) species as transient intermediates in several chemical transformations, we are planning to further investigate the electronic properties and reactivity of these complexes and their analogues and to explore their catalytic activity towards various C-C, C-N, C-O, and O-O coupling reactions involving Pd(III) intermediates, as well as investigate their small molecule activation reactivity.
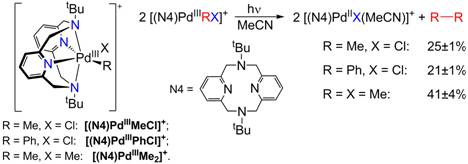
Figure 5. Stable mononuclear PdIII complexes and their light-induced reactivity.
