AmericanChemicalSociety.com
Reports: DNI7 48927-DNI7: Structural Relaxation of Polymers in Nanoconfined Geometries: Reconciling the Accelerated and Suppressed Physical Aging Observed in Different Systems
During the past 15 years studies of the physical aging of thin polymer films have frequently observed anomalous aging behavior relative to bulk materials with decreasing film thickness. The structural relaxation that occurs in non-equilibrium glassy systems leads to a decrease in free volume of the material over logarithmic time scales. Although these volume contractions are minuscule (<1%), the resulting property changes, termed physical aging, (e.g., mechanical properties and failure modes in particular) can be substantial and often adverse. As an example, the permeability of gas-separation membranes is exceedingly sensitive to the free volume in the polymer gas-sieving layer. Decreases in permeability of nearly 50% have been observed as a result of physical aging.
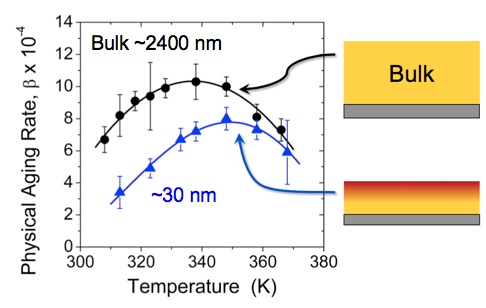
We have recently developed a new method of characterizing the physical aging rate of thin polymer films in an efficient manner using ellipsometry [1]. In comparison to other methods available for characterizing the structural relaxation of thin films, this method has the advantage that it can be easily applied to different polymers with varying chemical structure. The physical aging rate β is calculated from the time-dependent decrease in the film thickness: β = − d(h/h0) / d(log t), i.e., the slope of the data shown in Figure 1. Using this technique, we have measured the temperature dependence of the physical aging rate in ultrathin supported polystyrene (PS) films (Figure 2) and have found reduced physical aging rates at all temperatures for film thicknesses < 100 nm [2]. Our analysis of these results have demonstrated that the physical aging rates in these ultrathin films are not simply shifted in correspondence with the average glass transition temperature (Tg) reductions in these films, but that the reduced physical aging rates result from a gradient in enhanced dynamics present near the free surface of these films. The temperature dependence of the length scale characterizing the depth to which these enhanced dynamics penetrate into the film, from our analysis using a common two-layer model and a new gradient model, are in very good agreement with similar length scales characterizing the gradient in Tg dynamics, strongly suggesting that these two phenomena are related.
Figure 1
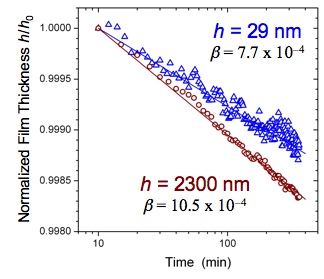
Figure 2
The most significant discovery that we have found thus far pertains to the physical aging rate of micron thick polymer films that are quenched in different geometries. We have found that the physical aging rate of micron thick polystyrene films quenched in a free-standing state exhibit faster aging rates with decreasing film thickness (accelerated aging), while supported micron thick films show no change in aging rate with thickness, as shown in Figure 3 [3]. This behavior is similar to that observed by Don Paul (U. Texas-Austin) and others in the gas permeation community. However, it is the first time that this behavior has been seen with a flexible C-C backbone polymer, in comparison to the high Tg, stiff polymers containing aromatic groups within the backbone that are traditionally used by the gas permeation community. Most illuminating is that we transfer our free-standing quenched films to a silicon support for the actual aging measurement. Thus, our results indicate that fundamentally much of the glassy state dynamics are controlled by the initial conditions present during the formation of the glassy state. The implications of this are that by manipulating the glass formation process, it may be possible to minimize some of the resulting physical aging behavior and improve film stability.
Figure 3
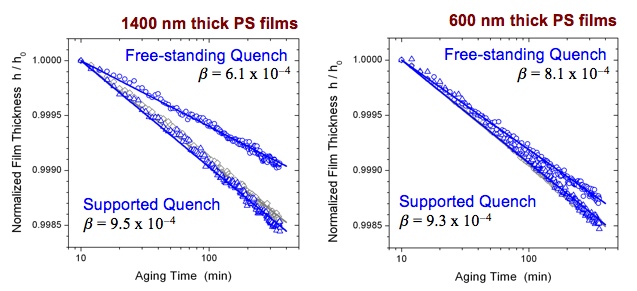
Recent studies in the research literature have demonstrated links between mechanical stresses and material deformation with physical aging rates and glassy mobility [4]. Because these accelerated aging effects shown in Figure 3 are occurring at micron film thicknesses, much larger than the < 100 nm thicknesses at which we observed the reduced aging rates that were correlated with Tg reductions [2], we believe this accelerated physical aging behavior to be unrelated to confinement effects. The observation that a film thickness dependence of the physical aging rate only occurs for films quenched in a free-standing, but not supported, state suggests to us that this is a bulk phenomenon where the material experiences different mechanical conditions imparted by the different film supports. Studies are underway to determine the specific cause of the film thickness dependence observed in Figure 3 on the physical aging rate by systematically varying the glass formation process to identify the key parameters affecting the aging process.
[1] Baker, E. A.; Rittigstein, P.; Torkelson, J. M.; Roth, C. B. J. Polym. Sci., Part B: Polym. Phys. 2009, 47, 2509-2519. "Streamlined Ellipsometry Procedure for Characterizing Physical Aging Rates of Thin Polymer Films"
[2] Pye, J. E.; Rohald, K. A.; Baker, E. A.; Roth, C. B. Macromolecules 2010, ASAP DOI: 10.1021/ma101412r. "Physical Aging in Ultrathin Polystyrene Films: Evidence of a Gradient in Dynamics at the Free Surface and Its Connection to the Glass Transition Temperature Reductions"
[3] Roth, C. B. Polym. Prepr. 2009, 50, 792-793. "Significance of Free-Standing vs. Supported Quench in Subsequent Physical Aging of Thin Polymer Films"
[4] Roth, C. B. J. Polym. Sci., Part B: Polym. Phys. 2010, DPOLY special issue (December). "Mobility and Stability of Glasses"
