AmericanChemicalSociety.com
Reports: B4 47629-B4: Pentacyclo[4.3.0.0(2,4).0(3,8).0(5,7)]non-4-ene: Synthesis, Reactivity, Matrix Isolation Spectroscopy, Calculations, and Physical Study of Reaction Products
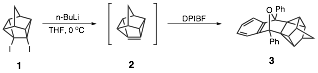
Undergraduate students in my research group have shown that treatment of 4,5-diiodopentacyclo[4.3.0.02,4.03,8.05,7]nonane (1) with n-butyllithium or t-butyllithium at 0 oC in the presence of a trapping agent such as 1,3-diphenylisobenzofuran (DPIBF) furnishes a 25-30% yield of the "trapped" product 3, thus providing strong evidence for the formation of 2.
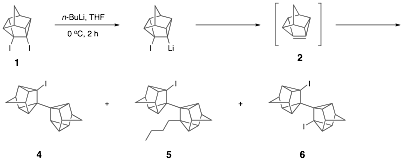
Dehalogenation of diiodide 1 with n-butyllithium in the absence of 1,3-diphenylisobenzofuran or
other trapping agents affords alkyllithium addition
products 4, 5, and 6 (and trace
amounts of other alkyllithium addition products). These three products were also present in the aforementioned
Diels-Alder trapping reaction of 1
with n-butyllithium
and DPIBF. Similar alkyllithium addition products are
formed when the dehalogenation is conducted with t-butyllithium.
It is well known that alkyllithiums add readily to highly pyramidalized alkenes
(see Tetrahedron 2005, 61, 5147), and in fact,
isolation of these addition products provides further evidence for the
formation of pyramidalized alkene
1.
In
order to further study the chemistry and reactivity of 2, an alternative route that does not utilize alkyllithiums
to generate the pyramidalized double bond must be
developed. Thus, one focus of our research during the grant period has been the
synthesis of these alternative precursors. An additional focus has been the use
of diiodide 1
to generate 2 without using alkyllithiums. In
his synthesis of cubene, Eaton observed that alkyllithiums reacted rapidly with
the pyramidalized double bond (see J. Am.
Chem. Soc. 1988, 110, 7230-7232). The high reactivity of cubene with alkyllithiums in the
reaction mixture prevented an exploration of the reactivity of the cubene
double bond with reagents other than alkyllithiums, a situation similar to what
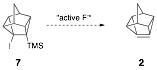
is observed in our synthesis of 2. Thus Eaton developed another route to cubene without using alkyllithiums
via the fluoride ion induced
elimination of 1-halo-2-(trimethylsilyl)cubanes. In a
similar manner, 4-iodo-5-(trimethylsilyl)pentacyclo[4.3.0.02,4.03,8.05,7]nonane (7) offers an alternative route to 2 via treatment with active fluoride
ion.
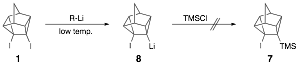
Previously,
attempts to synthesize 7 via
lithiation of diiodide 1 at low
temperature followed by addition of chlorotrimethylsilane (TMS-Cl), were
unsuccessful, as lithium iodide elimination from 8 was rapid even at low temperature; trapping with TMS-Cl could not
compete with elimination.
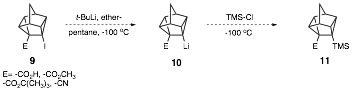
Moreover,
attempted lithiation of molecules of type 9 (E=-CO2CH3
,-CO2H, -CO2C(CH3)3, -CN), and
subsequent trapping with chlorotrimethylsilane also did not lead to molecules
of the type 11 (primarily) due to akyllithium addition to the electrophilic
E groups.
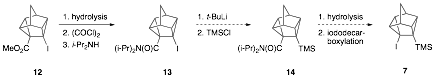
To avoid competition from
addition of alkyllthiums to the carbonyl and cyano groups present in 9, the N,N-diisopropylamide
13 was synthesized, as it is
relatively unreactive toward alkyllithium
addition to the hindered amide carbonyl. Currently, we are investigating metalation of diisopropylamide 13 with t-butyllithium followed by chlorotrimethylsilane quench to yield 14. Hydrolysis and Barton iododecarboxylation
should lead to 7.
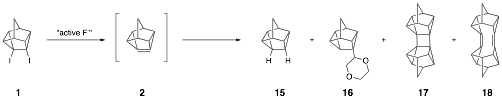
An alternative approach to 2 not utilizing alkyllithums
involves the dehalogenation of 2 with reactive metals such as sodium and potassium, and alloys
such as sodium amalgam. Other
groups have utilized this method extensively for the synthesis of pyramidalized alkenes. Thus, reaction of 4,5-diiodopentacyclo[4.3.0.02,4.03,8.05,7]nonane (1) with excess molten sodium in
refluxing 1,4-dioxane gave a mixture of four main
products by GC–MS. The product eluting first corresponded to the known
reduction product 15; the second product could be 16 (we have only crude NMR and GCMS at
this point), derived from the addition of the intermediate pyramidalized
alkene and the solvent (1,4-dioxane); the third
product has been identified by single
crystal x-ray analysis as cyclobutane
dimer 17;
the fourth major product has
the same m/z as 17 and is tentatively identified as diene
18.
As a result of the support
from this ACS-PRF grant, a total of six students have participated in this
project. Thus far, four of these students have presented posters at American
Chemical Society (ACS) National Meetings. Additionally, all of these students
have also presented at local and regional meetings such as the ACS Philadelphia
Section Poster Day and the Philadelphia Organic Chemists Club Poster Day.
The research described in this
proposal will continue to provide a current, in-depth and diverse research
experience to my students. Over the past ten years, I have mentored twenty-six
full-time undergraduate research students at Saint Joseph's University. These
students have gone on to seek advanced degrees in areas such as organic
chemistry, physical chemistry, inorganic chemistry, biochemistry, and molecular
biology. Other students currently seek and have completed medical degrees and
other professional degrees.
The funding of this project
has allowed all of the research students to be exposed to a variety of modern
organic synthetic techniques, and to routinely run reactions under an inert
atmosphere using Schlenk procedures and in an inert
atmosphere glove bag. The nature of this work will provide my research students
with not only a strong experience in synthetic organic chemistry, but also an
introduction to a broad range of spectroscopic methods, including NMR, FTIR,
and MS, and matrix isolation spectroscopy.