AmericanChemicalSociety.com
Reports: ND6 48890-ND6: Adsorption and Chromatographic Separation of Chain Molecules on Nanoporous Substrates
Abstract. Adsorption of chain
molecules on nanostructured surfaces and within nanoscale pores is the key
mechanism of chromatographic separation and characterization of polymers, which
is one of the most widespread analytical techniques in the petroleum and
petrochemical research and development. Chromatographic separation depends on
the different types of physico-chemical interactions between the macromolecule
and the surface of porous particles packed into a chromatographic column. The
most efficient separation is achieved at the critical point of adsorption (CPA)
that corresponds to a delicate balance between repulsive steric and attractive
adsorption interactions coupled with confinement effects, which allows for
characterization of macromolecules based on their chemical composition,
microstructure, and topology, independent of molecular weight. A better
understanding of the pore structure effects, such as pore size and shape, is
needed for a rational design of chromatographic columns, choice of porous supports,
and optimization of experimental protocols. The primary aim of the proposed
program is to gain a fundamental understanding of the physico-chemical
mechanisms of polymer adsorption in nanopores and the conditions of CPA based
on the development of new methods for
multiscale molecular simulations of real chromatographic systems. The
most important practical outcome will consist in the theoretical determination
of CPA conditions for molecular weight-independent elution of linear
homopolymers, as well as statistical and block copolymers, with the example of styrene-butadiene
systems separated on unmodified and modified silica substrates of different
pore structure in typical chromatographic binary solvents. In the future, we
envision the extension of this approach to more complex molecular
configurations.
Results for the reporting period. The main focus of the research was on
developing new simulation methods for modeling equilibrium adsorption and
dynamics of chain molecules in nanopores. The work was done in three
directions: MC simulation, Self Consistent Field Theory (SCFT) modeling, and Fokker-Plank
(FP) method.
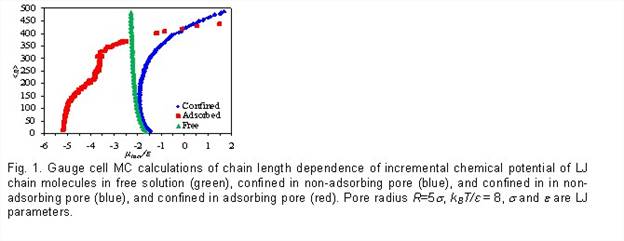
MC simulation (Chris Rasmussen, PhD student, and Dr. Aleksey Vishnyakov, Research Assistant Professor). We have implemented the gauge cell Monte Carlo method for non-branched chains of Lennard-Jones (LJ) atoms. We were able to obtain a quantitative agreement with the results obtained with modified-particle insertion method of Kumar et al 145 for free (non-adsorbed) chains. We have also simulated single chains in confinement, with and without adsorption potential, at various temperatures (Fig.1). We have found the gauge cell Monte Carlo method is an accurate and efficient technique for calculation of the free energies of polymeric molecules.
SCFT model for polymer partitioning between mobile and stationary phases. (Dr. Shuang Yang, postdoctoral fellow).
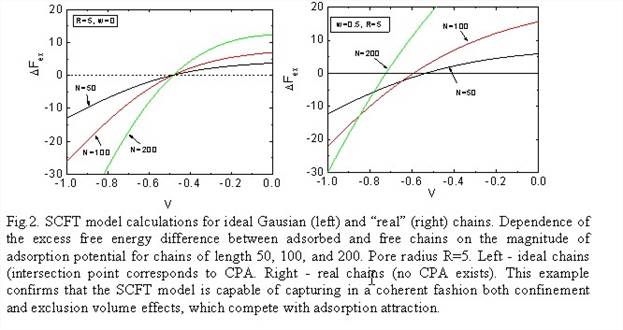
We developed a SCFT model for calculating the difference of the free energies of a macromolecule in mobile and stationary phases, which determine the equilibrium partition coefficient. SCFT represents polymer chain as a random walk trajectory in an external field. It is assumed that the chain is composed of Kuhn's segments (KS), or coarse-grained beads of size b, which depends on the chain stiffness. we performed instructive simulations in the most simple computational set-up to illustrate the feasibility of the proposed method. In Fig. 2, we present the excess free energy difference DFex(N,R) between confined and unconfined chains comprised of N = 50, 100, and 200 KSs for a spherical pore of radius R=5b. DFex(N,R) is plotted as a function of the magnitude adsorption potential, which was varied from 0 (SEC regime, no adsorption) to -1 (AC regime, strong adsorption).
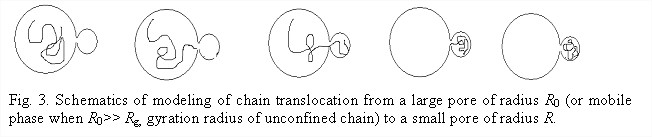
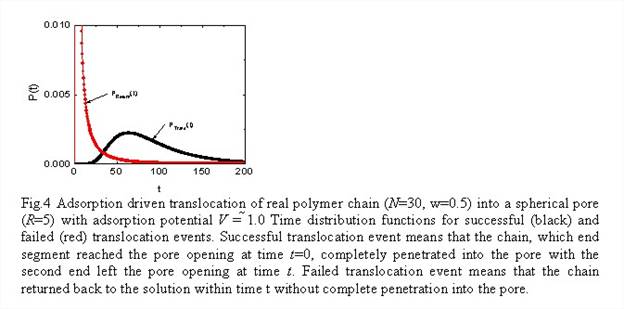
Fokker-Plank formalism for modeling dynamics of polymer adsorption (Dr. Shuang Yang, postdoctoral fellow). We adopted and advanced a FP formalism for modeling the dynamics of polymer adsorption in nanopores. As a case study system, we will first consider adsorption-driven translocation of a chain molecule from a solution to a spherical pore, Fig 3.
An instructive example, which shows feasibility of the FP method for studies of the dynamics of adsorption-driven chain translocation, is presented in Fig. 4.
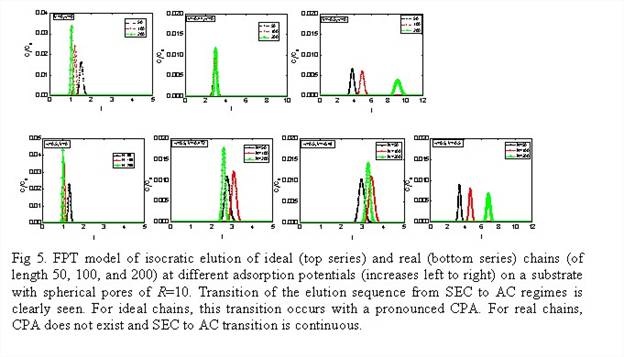
The results of thermodynamic simulations of partition
coefficients were employed in the first passage time (FPT) distribution model of
the elution time distribution. Our preliminary
results for a model system shown in Fig.2 are presented in Fig.5. The partition
coefficients were calculated with the SCFT model.
<>