AmericanChemicalSociety.com
Reports: AC1 48079-AC1: Asymmetric Halogen-Metal Exchange of Geminal Dihalides with Planar Chiral Organometallic Reagents
Introduction
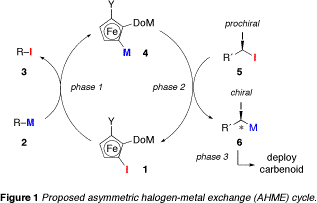
The goal of the funded research is to realize an asymmetric halogen-metal exchange (AHME) reaction of geminal dihalides using a recyclable planar chiral organometallic reagent derived from an iodoferrocene. The proposed AHME process (Figure 1) is a closed cycle in which a recyclable planar chiral ferrocene is used as a stereoinductive element. The complete AHME cycle incorporates three distinct phases: [1] generation of a planar chiral ferrocenylmetal (4) from an iodoferrocene precursor (1) via iodine-metal exchange using a simple achiral alkylmetal reagent (2); [2] AHME between 4 and a prochiral geminal diiodide (5) leading to the regeneration of 1 and the formation of a chiral a-haloalkylmetal (6); and [3] trapping of the scalemic carbenoid 6 in some process of interest.
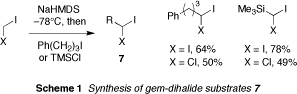
Results and discussion Geminal dihalide substrates for
study were prepared by alkylation of halomethyl sodiums with either TMSCl or a
1° alkyliodide via an adaptation of known methods (Scheme 1). For initial
assessment of the thermodynamic feasibility for the necessary halogen-metal
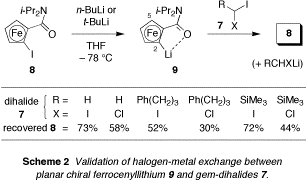
interchange reactions involved in AHME, a simple chiral 2-iodo-ferrocene (8) was prepared.
Following generation of the corresponding ferrocenyllithium 9 by I/Li interchange, a gem-dihalide was introduced
and the quantity of iodoferrocene 8
regenerated by the reverse Li/I exchange was determined (Scheme 2). These
results successfully verified that all of the requisite halogen-metal exchanges
were possible. In each case, interchange efficiency was greatest for the
prochiral geminal diiodides as compared to related geminal chloroiodides.
Interestingly, an analogous series of bromide based dihalides failed to
undergo halogen-metal exchange.
In our original design for
reagent 4 we considered that YH would
be important, in part because this attribute precludes a possible racemization
pathway via intermolecular enantiotopic proton transfer between metallated
ferrocenes. Fortunately, the fear of ferrocene racemization for carboxamide
based systems with Y=H proved to be unwarranted. A selection of the above
interchange cycles were repeated under otherwise identical conditions using an
enantioenriched sample of iodoferrocene 8 (96% ee); in no case was a measurable drop in the ee of recovered
material 8 observed. In addition to the simple
carboxamide based iodoferrocene 2, a
variety of other types of planar chiral ferrocenyl iodides were also targeted
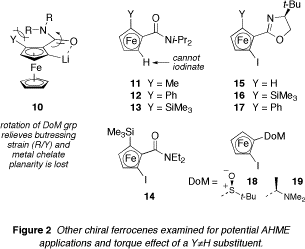
and tested for efficacy as potential AHME reagents (Figure 2). In many of these
compounds we tried to set YH. Aside from preventing possible racemization (vide
supra), another benefit of setting YH would be to allow for fine
tuning of the thermodynamic stability of the ferrocenyl metallate by a
combination of steric and electronic effects. For example, a butressing effect
between Y and the DoM group would result in torque and loss of chelate
planarity (as shown in 10). After
extensive experimentation, it was discovered that within the N,N-diisopropyl
carboxamide series, substitution at the Y position precluded installation of
the requisite iodine atom via DoM. Presumably, in this case the butressing
effect was too great and prevented DoM altogether. Within the analogous N,N-diethyl
series, however, synthesis of at least one iodide with YH (14)
was possible via DoM, albeit in a poor yield (14%).
An oxazoline based series of
ferrocenes (15-17), inspired by Sammakia's work and the FOP catalysts
of Overman, was also studied. In this case, iodoferrocenes with YH could be
prepared in high yield but the derived metallated derivatives (Li or MgCl)
proved incapable of effectively reassimilating iodine from dihalides 7. Sulfoxide 18 and tertiary amine 23 were also examined, but
again neither performed as effectively as carboxamide 8.
Imperative to a successful proof
of concept for the proposed AHME process is validation of the third and final
phase of the cycle: i.e., trapping of the product chiral carbenoid 6 in some useful reaction that could enable an
indirect assay of ee. To our immense frustration, early attempts to trap
carbenoid 6 with aldehydes
rarely yielded an addition adduct, but instead usually led to consumption of
starting dihalide and production of the alcohol arising from simple reduction
of the carbonyl. After exhaustive experimentation, it was discovered that such
carbenoids can be consistently added to carbonyl compounds in the desired
manner in the presence of Lewis acid additives. Our recent studies have also
demonstrated that low temperature quench following addition of a-chloroalkylmetals
to aldehydes leads exclusively to chlorohydrins, while analogous experiments
involving a-iodoalkylmetals
result in facile intramolecular collapse of the initial addition adduct to
provide epoxides exclusively.
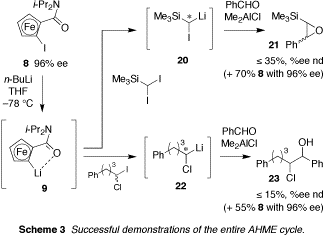
Complete AHME cycles have now
been successfully realized in two different scenarios (Scheme 3); these results
punctuate the culmination of two years of hard work on this difficult project.
Thus, enantioenriched 2-iodoferrocene 8
underwent initial I/Li exchange with n-BuLi at –78°C (THF);
following subsequent addition of gem-diiodide 7 (R=SiMe3, X=I), the resulting putative carbenoid 20 was thereafter added to benzaldehyde that had been
precomplexed with Me2AlCl. In this manner, epoxysilane 21 was obtained in 35% yield accompanied by 70% of
recovered 8 that exhibited no
significant loss in ee. In the second example, a similar sequence of events led
to chlorohydrin 23 (²15% yield)
by the use of racemic chloroiodide substrate 7 (R=Ph(CH2)3, X=Cl); again, 8 was recovered with excellent stereochemical
fidelity. The development of a suitable HPLC based resolution technique to
assay %ee for the products from these transformations is under active
investigation.
Conclusion In summary, validation of all
three phases of the proposed AHME cycle has been achieved. It was further
established that a simple planar chiral 2-iodoferrocenyl carboxamide reagent (8) could be subjected to a round trip I/Li/I sequence
without compromising its ee, thus allaying concerns of a possible racemization
pathway. Finally, a reliable method for the trapping of alpha-haloalkyl metals
with aldehydes was developed. It remains to assay ee for products resulting
from this process, to optimize the methodology, and to study and expand on its
potential scope. This work will be conducted and concluded during the upcoming
12 month no cost extension period (9/1/10-8/31/11).
Note: Research findings have been
presented by student Christopher Emerson at the 239th ACS National Meeting
(3/21/10-3/25/10, San Francisco) and at the 1st Annual Graduate Research
Symposium of the Organic Division of the ACS (7/15/10-7/18/10, Boston).