AmericanChemicalSociety.com
Reports: ND9 48919-ND9: Coarse-Graining of Molecular Dynamics Simulation via Model Reduction with Application to Polyolefin Tacticity for Morphology Control
Introduction
Intermediate order, which occurs in many polymers, e.g. polyvinyl chloride (PVC) and poly(n-butyl methacrylate) as well as specialty polymers such as poly(trimethyl silyl propyne), and poly(norbornene), is a structural order that is in between the crystalline and amorphous states. Polymers that show intermediate order have potential applications including membrane separation, photolithography and catalyst substrates. The initial object of our study is on PVC, where intermediate order produces a unique thermoreversible gelation property that makes PVC easier to process than its crystalline counterpart.
In general the computational capability does not allow us to evolve enough to reach the desirable state, e.g. intermediate order, where we can analyze the system properties, so we seek a model reduction approach to reduce the simulation burden.
Methods for coarse-graining of molecular simulations, by grouping nearby atoms, have been developed to speed up molecular dynamics simulations, but due to the complexity of polymer tacticity, the size of the groups has been limited to one monomer. However, much greater speedup is still needed. An automated method for model reduction and system identification could provide a complementary approach for computational reduction, such as equation-free computing. However, identifying an appropriate reduced-order state for equation-free computing remains a challenging problem, even for a simple fluid.
In our work we propose the modeling method for molecular dynamics simulations of polymers, which combines features from equation-free computing with polymer coarse-graining to benefit our study on the intermediate order of PVC.
Polymer Chain Building and WAXD
Intermediate order is detected using wide angle x-ray diffraction (WAXD). WAXD is an X-ray diffraction technique that is often used to determine the crystalline structure of polymers. We have constructed numerous PVC systems, each initialized as an ordered array of aligned chains. The basis of all of these systems is the unit cell for syndiotactic PVC as proposed by Hobson and Windle (1993), which we replicated in all directions.
Using the resulting general row and column arrangement, we made chain substitutions, exploring the ability of isotactic or atactic chains to fit themselves into the ordered syndiotactic environment. We tried several variations on this theme substituting in different percentages of purely isotactic and atactic chains, using different conformations of the isotactic chains, and constructing heterogeneous tiled systems. In each case, the system was subjected to a process of potential minimization and molecular dynamics. Then, we generated a simulated WAXD for the system, comparing it with experimental WAXD curves.
The purely syndiotactic system yields a simulated WAXD that contains the major features found in experimental WAXD curves for PVC. But as expected, we have observed that the greater the percentage of isotactic and atactic chains within the system, the further the simulated WAXD deviates from experimental findings. We are attempting to identify the physical structural features responsible for the peaks observed in the experimental WAXD pattern, the idea being that with that understanding, we will be able to incorporate isotactic and atactic chains without disrupting those features perhaps even enhancing them. We believe the major peaks of the experimental WAXD curves to be attributable to interchain interactions, and are now in the process of sorting out the geometry of those interactions.
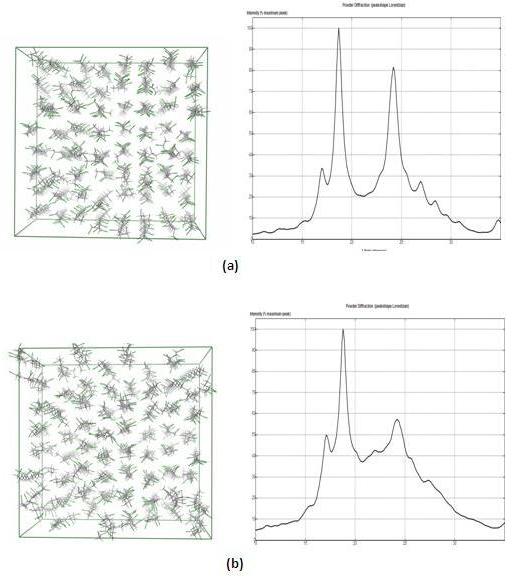
Figure 1. (a) 100% syndiotactic system and its corresponding simulated WAXD curve. (b) 50% syndiotactic, 50% isotactic system and its corresponding simulated WAXD curve.
Approach
We propose an approach where a modified local feature analysis is applied to molecular dynamics simulations, to automatically identify seed atoms, based on correlations in the dynamic motion of the atoms. Unlike typical coarse-graining methods, the atom groups defined by these seeds may or may not be on the same polymer chain. We then apply time-series techniques to propagate forward the trajectories of the seed atoms into the future time T. The positions of all atoms can then be reconstructed via simple matrix transformation. We define this process, from atomistic simulation to the recovery of the system at time T, as one model iteration. At each iteration, the local feature analysis is repeated, such that the choice of seed atoms may change over time as the polymer conformation changes. This would overcome the under-sampling problem caused by limited data collection from slow process.
Only simple matrix calculations are required to calculate the local feature analysis and its reconstruction, so that the primary computation is associated with the short molecular dynamics simulations run at each step.
Simulation Example
The proposed approach has been applied to PVC system simulated in Molecular Operating Environment. Sixteen syndiotactic PVC chains with 8 monomers each were built and simulated in atomistic scale under NPT for 250ps. The conformation and WAXD curve of the system at the initial and end time are given in Figure 2. At the initial time, the polymer chains are well aligned while at 250ps they are more disordered. The corresponding WAXD at these two points are obviously different.
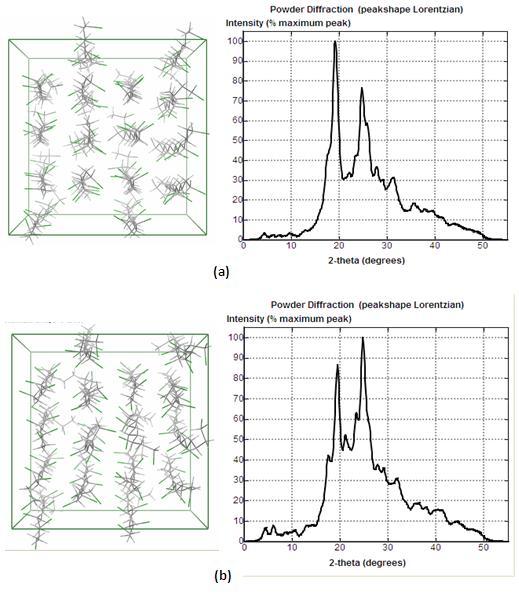
Figure 2. (a) Conformation and WAXD at initial point. (b) Conformation and WAXD at 250ps.
We studied scenarios where the ratios of detailed MD simulation time over the extrapolation time are 150ps/100ps and 100ps/150ps. That is, 150ps or 100ps detailed information are used for derive the seed atoms and mode the time trajectories while the remainder data are used to compare with the extrapolation results. The results are shown in Figure 3. With the proposed coarse graining and extrapolation method we recover the system conformation and WAXD curves very well. The 150ps/100ps scenario provides better accuracy compared to the 100ps/150ps scenario but also has longer training time.
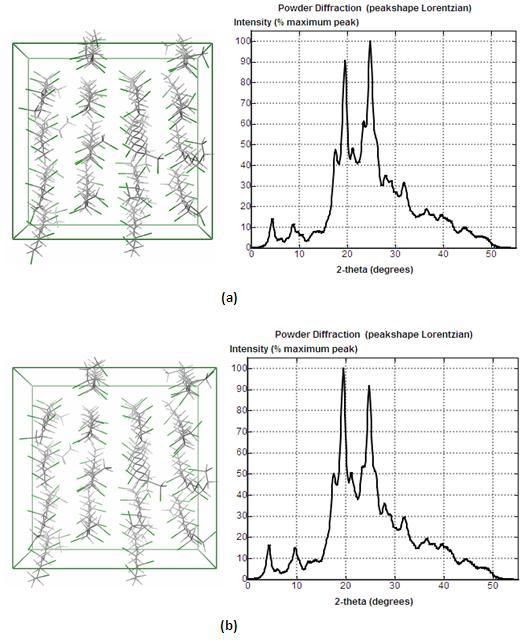
Figure 3. Conformation and WAXD for the recovered systems: (a) 150ps/100ps (b) 100ps/150ps.
Ongoing Work
Moreover, we are studying more advanced time series identification approaches, so we can predict longer term dynamics from shorter training period accurately. The ultimate goal of this research is to use the proposed algorithm to formulate mesoscale models that retain the dynamics and physics responsible for the intermediate order, and to use such models to explore the formation of intermediate order in new polymers.
