AmericanChemicalSociety.com
Reports: DNI3 48999-DNI3: Can Vanadium Pyrophosphate Coordination Complexes Perform Butane Oxidation?
Critical results obtained as part of the PRF project are highlighted below. We have been able to make significant progress during the first year of the project and are well set-up to translate this into a successful second year. The grant has allowed me, along with support from Syracuse University (who funded a post-doctoral student) to establish a strong Pyrophosphate group at SU. In addition, I was recently tenured, aided in part by this award. A new collaboration was established in between a catalytic group in New Zealand and my graduate student Amanda Hoffman will travel to New Zealand to conduct VPO research in 2011. This is due to the initial support of this project by the ACS PRF fund and will have a hugely positive impact on her young career, while also cementing a strong connection between the two labs moving forward.
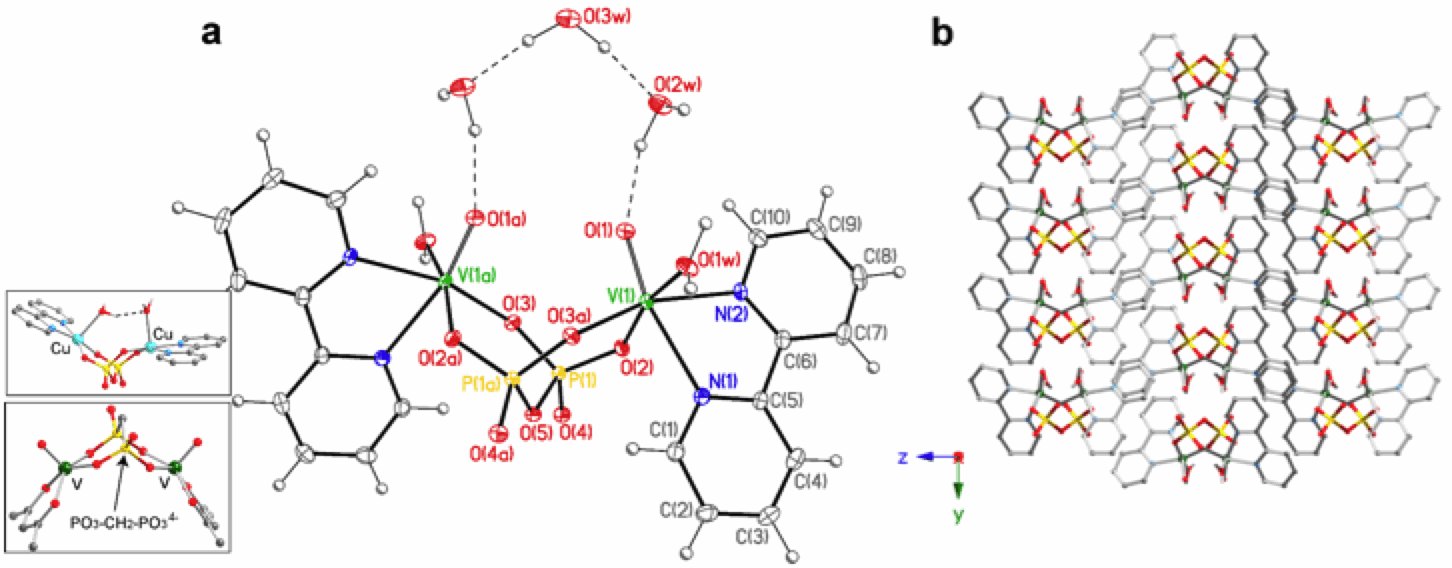
We have discovered that by reacting vanadyl salts such as VOSO4 or VO(acac)2 with PPi in the presence of 2,2'-bipyridine (bipy) as capping ligand, a dimeric complex can be formed at room temperature, performing a standard 'in solution' reaction. This material is highly insoluble and readily precipitate out of solution (H2O or a mixture H2O/alcohol) as a fine powder. Although investigation through single crystal X-ray diffraction was prevented by difficulties in attaining suitable crystals initially (see below), its dinuclear structure was elucidated from elemental and thermogravimetric analysis (TGA), magnetic susceptibility measurement, IR spectroscopy, and microanalytical data, all consistent with the formulation [VO(bipy)]2(μ-P2O7)]. The TGA profiles indicated the loss of five water molecules of crystallization between 25–140°C, followed by sample decomposition over 300°C. Structural information were extrapolated from the IR spectrum, which was almost superimposable with that of the known Cu(II) compound {[Cu(bipy)(H2O)]2(µ-P2O7)}á7H2O (see insert in Figure 1), the major divergence between the two being the occurrence of a band centered at 972 cm-1 for the vanadyl compound, as expected (V=O stretching frequency). A possible formula {[VO(bipy)]2(µ-P2O7)}á5H2O and a molecular geometry similar to that of the Cu(II) dimer (penta-coordinated VO(II) ion) was thus theorized. Single-crystal structural data was highly sought after then to better understand the pronounced insolubility, at odds with the solubility of the Cu(II) analogue.
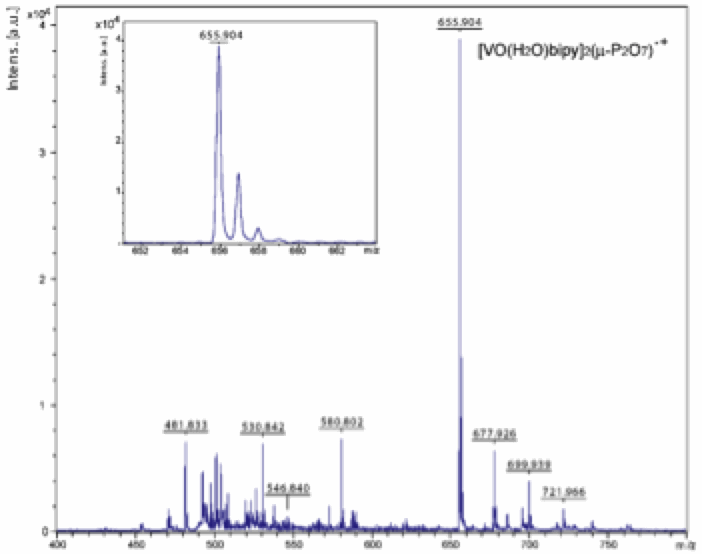
We have recently accomplished this goal, with the achievement of the crystal structure of the first vanadyl-PPi complex (see Figure 1; unpublished data). IR, elemental and TGA analyses of the crystals grown with slow diffusion techniques perfectly match with analyses performed on the fine precipitate from the direct synthesis. Single-crystal X-ray diffraction data (100 K) agree with the preliminary results analysis/interpretation, confirming both the nuclearity of the complex (as dimer) and the solvent content, while insolubility is observed to derive from classical supramolecular interaction such as hydrogen bonds (as already reported by DuPont) and ¹-¹ stacking (no V=O∙∙∙∙∙V=O interactions). Quite unexpectedly, two of the five water molecules in the crystal are coordinated to the metal, one to each VO(II). As a consequence, the VO(II) ions are hexa- and not penta-coordinated as originally supposed (and as observed, for instance, in the acac, methylene-bisphosphonate analogue obtained under hydrothermal conditions, see insert in Figure 1). This result suggests that water is a good coordinating solvent for VO(II) (as well as for Cu(II)) and thus not ideal for the formation of penta-coordinated VO(II) species. MALDI-TOF analysis of the reaction solution confirms the occurrence of the same hexacoordinated species in both crystalline phase and solution (Figure 2).
Pyrophosphate adopts the classical bridging mode, separating the two metal ions by~ 4.5 . The two VO(II) units in the dimer are syn-oriented, with a dihedral angle of 29° (only 8° for the cis-Cu-OH2 bonds in the Cu(II) analogue) and a O∙∙∙O separation of ~ 3.2 , which perfectly matches with the (parallel) VO(II) units separation in the structure of VPP.
Figure 1. Crystal structure of the VO(II)-PPi
dimer of formula {[VO(bipy)(H2O)]2(µ-P2O7)}á3H2O
(a) ORTEP plot (30%) [(a) 2-x, y, 1.5-z)];
(b) view of the crystal packing
along the a axis showing extensive
¹-¹ interactions between the bipy ligands (crystallization water molecule
omitted for clarity). The inserts shows the structure of our Cu(II)-PPi-bipy
analogue and of the methylene-phosphonate (MePP) analogue [VO(acac)]2(µ-MePP). This may be extremely important for catalysis
since we can, in theory, gradually de-hydrate the crystals and make the two
vanadyl units available for interaction with the substrate.
Figure
2. MALDI-TOF spectrum of the
reaction solution showing a peak 656 m/z consistent with {[VO(bipy)(H2O)]2(µ-P2O7)}+,
with the theoretical corresponding isotopic distribution also observed (inset). Given the
potential held by this structure in terms of catalytic applications, we have
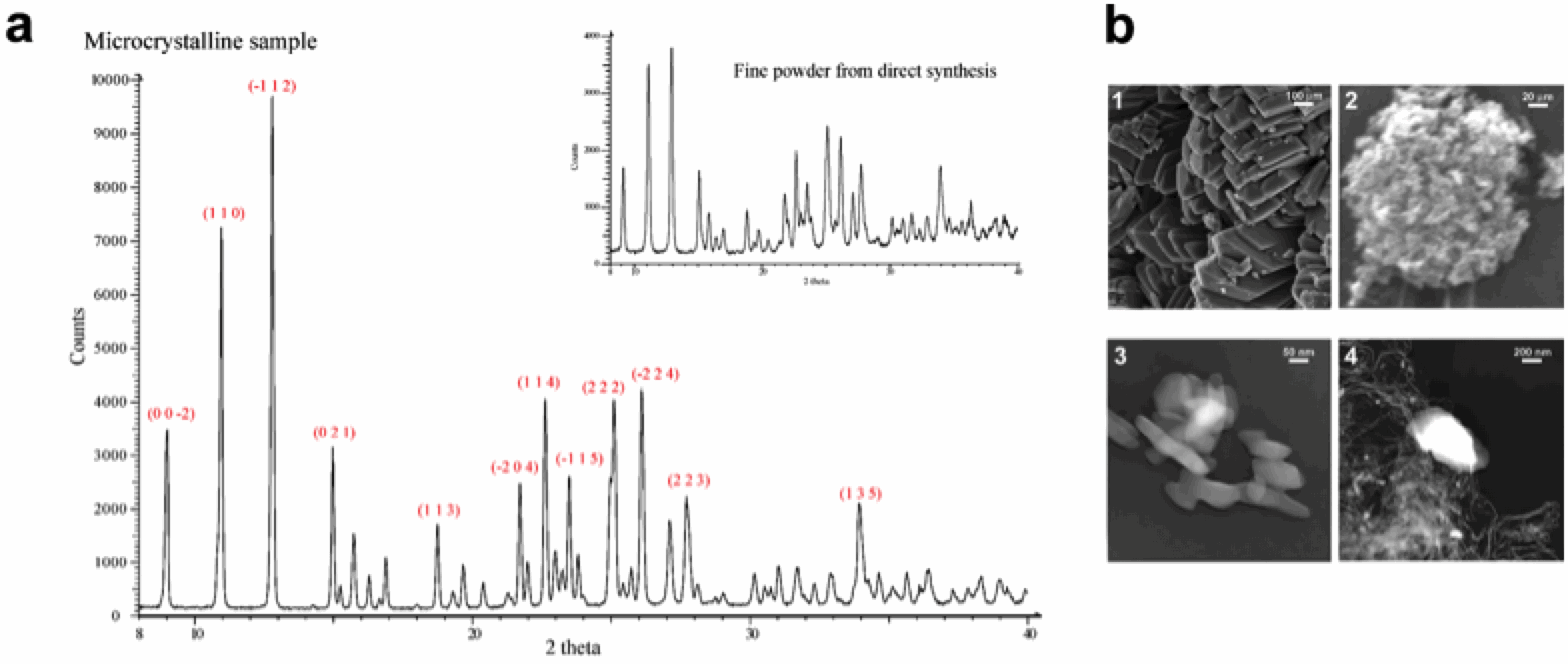
further investigated the bulk phase, via
powder X-ray diffraction (XRD) and electronic microscopy (SEM/TEM). XRD
profiles of both the crystalline (slow crystallization process) or powder
(precipitated) bulk phases. Both phases have excellent agreement with the
theoretical spectrum calculated from our single-crystal diffraction data
(Figure 3a). The microcrystalline nature of the powder was further confirmed by
TEM, with the occurrence of nano-plates resembling the single-crystals detected,
see Figure 3b.
Figure 3. (a) Experimental powder diffraction
pattern [2 theta range 8-40°, CuKα] recorded at room temperature for
micro- and nano-crystalline samples of {[VO(H2O)bipy]2(μ-P2O7)á3H2O.
(b) SEM (top) and TEM (bottom) images of micro- (1 and 3) and nano- (2 and 4)
crystalline samples of {[VO(H2O)bipy]2(μ-P2O7)á3H2O.
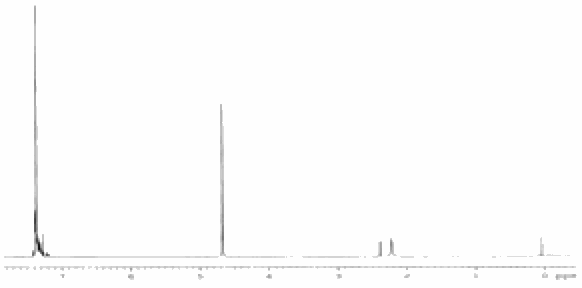
A preliminary investigation of
the catalytic activity of this complex in the presence of oxygen using butanol
as substrate shows 100% conversion to the oxidized product, butanoic acid (see
Figure 4). Control experiments where sodium pyrophosphate was added showed no
conversion. This catalytic conversion based on a vanadium pyrophosphate 'core'
synthesized at room temperature is unprecedented. Catalytic tests with this
material embedded in support silica are underway as well as attempts to
crystallize other VO(II)-PPi structure with different {(VO)(P2O7)}
cores.
Figure 4. 1H NMR of (a) butanol and (b)
butanoic acid after conversion over vanadium pyrophosphate-bipyridine. Reaction
was conducted at 80 °C
over 1 hour in liquid butanol under a positive pressure of pure oxygen.
Approximately 600 turnovers were calculated. Note the complete disapperance of
the butanol methylene signal at 4.7 ppm.
a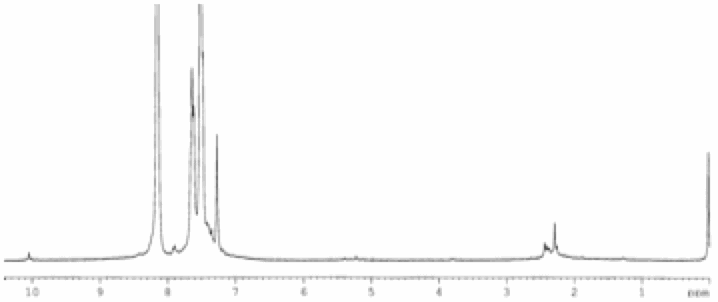
b
