AmericanChemicalSociety.com
Reports: DNI3 49646-DNI3: New Catalysis with Nickel Pincer Complexes
Development of catalytic reactions with inexpensive transition metals is becoming more important than ever as the costs of precious metals have risen and the natural reserves of these metals have declined. Although palladium remains to play a prominent role in modern homogeneous catalysis field, there is growing urgency to replace expensive palladium catalysts with much cheaper nickel catalysts for the sustainable development of the chemical industry. Our earlier studies focused on designing nickel-catalyzed reactions have led to the discovery of efficient and chemoselective hydrosilylation of aldehydes catalyzed by diphosphinito (PCP pincer) nickel hydrides. Because the fundamental chemistry of nickel pincer complexes is less developed compared to palladium pincer complexes, and because the steric and electronic properties of the nickel center can be readily tuned with the versatile pool of pincer ligands available, there are great opportunities and potentials for the development of new catalysis with nickel pincer complexes.
For the first year of the project, our research has been focused on the mechanistic investigation of various insertion reactions with nickel hydride complexes. We have shown that CO2 inserts readily into a nickel hydride supported by a diphosphinito pincer ligand (1, in Scheme 1). Isotope-labeling experiments have suggested that this insertion reaction is reversible, with the equilibrium constant favoring the nickel formate 2. We have also studied the kinetics of the deinsertion of CO2 from 2 by using CS2 to trap the hydride intermediate to make the more stable CS2 insertion product. When a large excess of CS2 is employed, the overall reaction rate is independent of the CS2 concentration, thus providing us with the information on the first-order deinsertion step. The obtained activation parameters are consistent with DFT calculations carried out by our collaborator (Prof. Kuo-Wei Huang from KAUST). The mechanism of CO2 insertion is more likely to proceed via a direct hydride transfer process, rather than involving the coordination of CO2 to the nickel center.
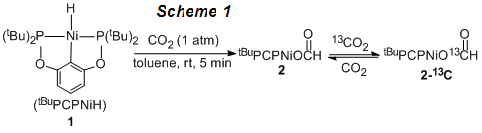
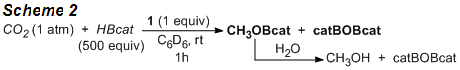
Progress has also been made toward the development of catalytic reactions based on our mechanistic studies. We have reported that the reaction of 2 with catecholborane (HBcat) yields a methanol derivative, which has been supported by NMR and mass spectral analyses. Further investigations have led to the development of catalytic reduction of CO2 with HBcat in the presence of only 0.2 mol% of 1 (Scheme 2). Following the hydrolysis of the initial reduction product, methanol can be obtained in 61% yield.
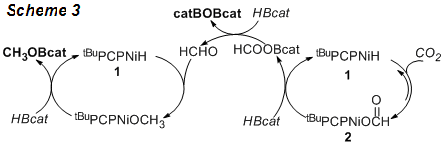
Catalytic cycles consistent with our observations have been outlined in Scheme 3. The catalytic reduction of CO2 begins with a reversible insertion of CO2 into a Ni-H bond. The subsequent cleavage of Ni-O bond with HBcat regenerates 1 and releases HCOOBcat, which is reduced to formaldehyde by another HBcat. A second cycle involves the hydroboration of formaldehyde, which is analogous to the nickel-catalyzed hydrosilylation of aldehydes reported earlier. In accord with this mechanistic hypothesis, the reduction of paraformaldehyde with HBcat in the presence of 1 also gave CH3OBcat. Finally, the independently synthesized 2 exhibited comparable catalytic efficiency as 1, an advantage in our catalytic system as compound 2 is air stable.
In summary, we have revealed important mechanistic insights into CO2 insertion into a nickel hydride complex, and we have shown the first catalytic hydroboration of CO2, with the highest turnover frequency (495 h-1 based on B-H) reported to date for the reduction of CO2 to the methoxide level. Our further studies will be focused on elucidating more mechanistic details, improving the catalytic efficiencies, utilizing inexpensive reducing reagent (such as H2), and developing other related catalytic reactions.
