AmericanChemicalSociety.com
Reports: B6 47856-B6: The Development of Accelerated Molecular Dynamics for Complex Gas-Phase Reactive Systems
A suite of programs called Accelerated Molecular Dynamics with Chemistry (AMolDC) has been written, tested, and employed in order to perform adaptive, multilevel QM/MM simulations for complex chemical processes in the gas-phase. During the last year a paper that examines the properties of this new method was published.1 The method is formulated to give a time-dependent, multilevel representation of the total potential that is derived from spatially-resolved quantum mechanical (QM) regions. A 110 atom system was used to demonstrate the continuity and energy conserving properties of the method. The effect that a discontinuous total potential resulting from the adaptive nature of the total potential has upon the total energy and the kinetic energy of the system was examined. In addition, the effect of the discontinuities in the magnitude of atomic force vectors due to changing the electronic structure during the simulation was examined, as well as the effect that these discontinuities have upon the atomic kinetic energies. The method was shown to exhibit smooth continuous atomic kinetic energies, even for those atoms involved in a change of groups and, thus, QM theory used to describe them. Thus, the method, while not conserving total energy, does yield canonical (NVT) simulations. In addition, the time-reversibility property of the simulation with an extremely discontinuous total potential was also examined. Lastly, the computational scaling associated with formulating the total potential as a multi-body expansion in terms of spatially-resolved groups was investigated. Ref. 1 shows that the AMolDC method scales linearly with system size due to the fact that at a constant temperature and pressure, the average system size will remain approximately constant regardless of the number of atoms in the simulation.
The last year of work has been centered on several items: i.) linking several external QM and MM codes to AMolDC ii.) writing a more robust version of AMolDC that may submit the QM jobs in serial or parallel iii.) examining the computational scaling of AMolDC in both serial and parallel, iv.) writing the database that the QM data are saved to that is easily expandable as the simulation proceeds, v.) writing the interpolation module that will use the saved grid of QM data for a given group to perform fast and accurate numerical interpolations instead of another QM calculation. Each of these is examined in turn.
| Fig. 1 Computational scaling of the serial version of AMolDC. |

First, the work of Ref. 1 examined the properties of AMolDC as an adaptive, multilevel QM methodology because at the time linking the code with an empirical molecular mechanics (MM) force field hadn't been accomplished. This work has now been finished by linking the code to the Reax force field.2 In addition, AMolDC can perform QM calculations using GAMESS,3 PSI3,4 and NWChem.5 The multiple external QM codes ensure that AMolDC is capable of using many different types of QM theory, i.e., from low level HF and DFT methods to high multi-reference CAS-SCF and couple cluster (CC) calculations.
Second, the CalcPot module of AMolDC was originally written to submit the QM and MM jobs in serial. CalcPot was rewritten to be capable of using distributed computing resources should they be available. Now, as part of the input deck for AMolDC, a variable is defined that gives the number of compute nodes that are available for running QM jobs and all the QM jobs are distributed across these nodes.
| Fig. 2 Computational scaling of the parallel version of AMolDC. |
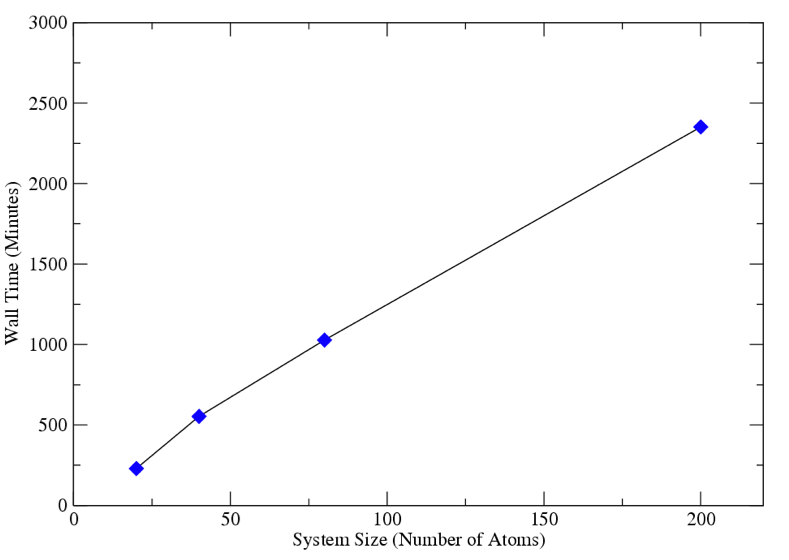 Third, the computational scaling of the QM/MM
version of AMolDC was examined in both serial and in parallel. Contained in figures 1 and 2 are the
total wall time for running 1000 time steps of various system sizes of H2
at 400 atm pressure and a temperature of 1000 K. In Fig. 1, the QM calculations were performed with at the
HF/6-31g** level of theory and in Fig. 2 at the HF/cc-pVTZ level of theory and
distributed across 6 compute nodes.
As can be seen in Figs. 1 and 2, AMolDC utilized in both serial and
parallel, yields linear scaling as a function of system size.
Third, the computational scaling of the QM/MM
version of AMolDC was examined in both serial and in parallel. Contained in figures 1 and 2 are the
total wall time for running 1000 time steps of various system sizes of H2
at 400 atm pressure and a temperature of 1000 K. In Fig. 1, the QM calculations were performed with at the
HF/6-31g** level of theory and in Fig. 2 at the HF/cc-pVTZ level of theory and
distributed across 6 compute nodes.
As can be seen in Figs. 1 and 2, AMolDC utilized in both serial and
parallel, yields linear scaling as a function of system size.
The fourth and fifth accomplishments of the year have taken a tremendous amount of time to write and test the various programs needed to perform the tasks of saving the QM grid of data and use these data as a basis for local interpolations. The potential energy surface (PES) database has to be easily extendable to very diverse groups, capable of growing as new groups are formed, and quickly accesses to retrieve the stored QM data. The interpolation module must be capable of performing very fast and accurate interpolations over very diverse QM grids and groups. Some groups may always be around minima (non-reactive groups), while some may have bond distances that get large (reactive groups). The modules have been written and computational scaling and accuracy tests are currently being performed. Initial results show that the combination of the database and interpolation modules are saving a factor of 3 in computational expense for a very small three atom H2O system treated at the SCF/6-31G level of theory.
References:
1. Guthrie, M.G.; Daigle, A.D; Salazar, M.R. J. Chem. Theory Comput., 2010, 6, 18.
2. Chenoweth, K; van Duin, A.C.T.; Goddard, W.A. J. Phys. Chem. A, 2008, 112, 1040.
3. M.W.Schmidt; K.K.Baldridge; J.A.Boatz; S.T.Elbert; M.S.Gordon; J.H.Jensen; S.Koseki; N.Matsunaga; K.A.Nguyen; S.Su; T.L.Windus; M.Dupuis; J.A.Montgomery; J. Comput. Chem., 1993,14, 1347.
4. T. Daniel Crawford; C. David Sherrill; Edward F. Valeev; Justin T. Fermann; Rollin A. King; Matthew L. Leininger; Shawn T. Brown; Curtis L. Janssen; Edward T. Seid; Joseph P. Kenny; and Wesley D. Allen, J. Comp. Chem. 2007, 28, 1610.
5. D. E. Bernholdt; E. Apra; H. A. Fruchtl; M.F. Guest; R. J. Harrison; R. A. Kendall; R. A. Kutteh; X. Long; J. B. Nicholas; J. A. Nichols; H. L. Taylor; A. T. Wong; G. I. Fann; R. J. Littlefield; J. Nieplocha, Int. J. Quantum Chem. 1995, 29, 475.
