AmericanChemicalSociety.com
Reports: AC3 48022-AC3: Late Transition Metal Complexes with Fluorinated Alkoxide Ligands for C-H Activation
We proposed the studies of a new family of late first row transition metal complexes having oxygen donor ligands in low, medium and high oxidation states with fluorinated alkoxide oxygen donor ligands. Our goals were (i) preparation of new transition metal complexes with convenient oxidation states, (ii) preparation of low and high oxidation species without additional ligands, (iii) preparation of high oxidation state complexes via atom transfer reactions, (iv) placement of oxygen donor ligands such as OC4F9, OCH(CF3)2 and related species in the ligand field series, and (v) to survey these new reactive systems for development into catalytic C-H oxidation systems under mild conditions. Last year we accomplished goals (i) and (iv), and this year we have made progress further progress on (ii) and have some interesting observations with regard to objectives (iii) and (v).
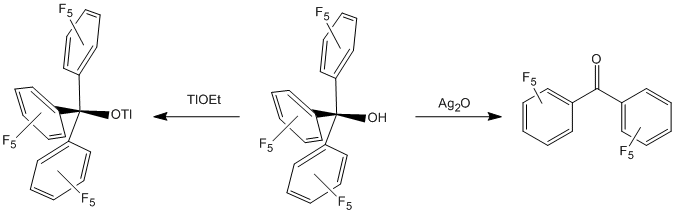
Although the starting materials [M(OC4F9)n]m- were readily prepared in crystalline form, the oxidized neutral products (M = Ni, Co) have not been isolated except as oils. Therefore, a tertiary alcohol with extensive fluorination but fewer degrees of rotational freedom was required and investigations were begun on a related tertiary alkoxide ligand, -OC(C6F5)3. The parent alcohol was described in the literature but primarily in the studies of trityl cation analogs. The compound is prepared by reaction of LiC6F5 with perfluorobenzophenone and hydrolysis of the resulting lithium alkoxide with water, to which we have added an X-ray crystallographic study.
Reactivity patterns of HOC(C6F5)3 were not entirely as expected based on our published work with OC4F9 and fluorinated aryloxides. The alcohol was readily converted to TlOC(C6F5)3 via TlOEt, but use of Ag2O as an oxide base source toward the putative AgOC(C6F5)3, led to black powder (presumed Ag0) and the ketone from which the alcohol had been prepared. Several attempted metathesis reactions of transition metal halides MXn with TlOC(C6F5)3 were consistent with precipitation of the highly insoluble TlX, but these reactions also formed copious quantities of perfluorobenzophenone as evidenced by 19F NMR and ESI-MS spectroscopies. Undaunted, we pushed on and tested two alternative synthetic approaches to [M{OC(C6F5)3}n]m- target compounds.
Bonding
of this ligand to transition metals has been pursued
through two routes: (i) alcoholysis
of strong internal bases in complexes such as [M{N(TMS)2}2]
and {Cu(mes)}n and (ii) reaction of metal
halides MXn with the alcohol in the
presence of Et3N. The former
route has been successful in the preparation of a microcrystalline purple
powder whose elemental analysis is consistent with {Co(OC(C6F5)3)2}-0.5
C6H14. Structural
data have not been obtained but we posit a bridged dimeric
structure of the form [(F5C6)3CO-Co-(m2-OC(C6F5)3)-Co-OC(C6F5)3]
based on literature work with the bulky tri(cyclohexyl)methanol. Preliminary work with this Co(II)
species shows reactivity with both oxidizing and reducing agents, and work is
underway vigorously to characterize the putative Co(III) and Co(I) products, as
well as pursue two-electron oxidation chemistry of the Co(I) derivatives. Expanding our work to bidentate,
chelating ligands, we have greatly increased the
little chemistry that was known with the dodecafluoropinacolate
(ddfp) ligand. Preliminary work on combination of H2ddfp
with transition metal sulfates, with an aqueous solution of NaOH
and the alcohol. Prior to our work only
the structure of [K2Ni(ddfp)2]-4H2O
was known in the late metals and two Cr(V)(ddfp)
compounds had also been prepared. The Ni(II) compound is square planar and diamagnetic, showing
the relatively strong ligand field imposed by the ddfp ligand. We have now expanded this series to include
the paramagnetic Mn(III), Co(II) and Cu(II) in
{K(THF)2(18C6)}[Mn(THF)(ddfp), {K(DME)2}2[Co(ddfp)], [K2Cu(ddfp)2]-4H2O,
and {K(18C6)}2[Cu(ddfp)2]- 5 H2O. The monoacids (KHddfp),
{K(18C6)}(Hddfp), and
diamagnetic Cu(I) derivative [KCu(ddfp)(PPh3)]2-THF
have also been prepared and characterized structurally. The Cu(II) and Mn(III) compounds are air-stable, but the Co(II) and Cu(I)
species must be handled in anoxic environments.
We began our studies of lower valent transition metal species with Cu(I), and have
prepared several different derivatives including heterobimetallic
K and Tl complexes of the form {MCu(OR)2},
the related bis(alkoxides)
with weakly coordinating cations {K(18C6)}+
and (Ph4P)+, as well as the dimeric
phosphine compounds {(Ph3P)Cu(OR)}2. The synthetic pathways and highlights of the
characterization data collected to data are summarized in the scheme
below. Arguably the most important and prevalent redox chemistry of Cu(I) is with O2,
whether it be oxidase, monooxygenase,
or dioxygenase chemistry. Therefore we began our pursuit of reactive
and useful high oxidation states by mixing O2 with the most simple Cu(I) derviatives. These {KCu(OR)2} compounds showed immediate reactivity with
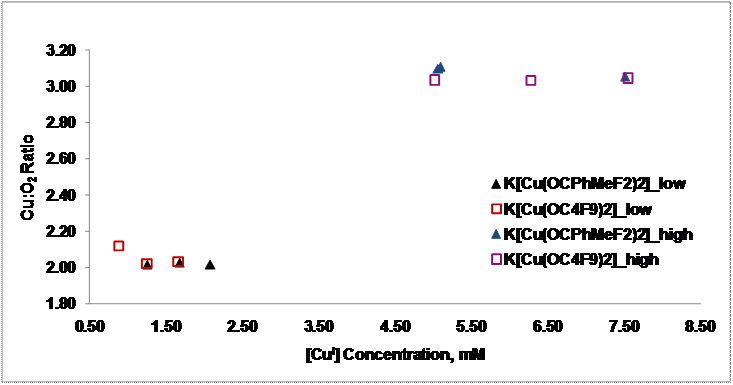
O2 at -78°C. Interestingly, the observed chromophores are dependent on the relative Cu(I) concentration in solutions that are saturated with O2. Manometry studies
showed that for Cu(I) concentrations less than ~ 2.0 mM the Cu:O2 ratio is 2:1 but for concentrations
above 4.5 mM, the ratio is 3:1.
Furthermore,
the {KCu(OR)2}
complexes do not behave as 1:1 electrolytes in either THF or CH2Cl2,
the former being particularly surprising.
The complexes with weakly coordinating cations
{K(18C6)}+ and (Ph4P)+
in A+[Cu(OR)2]- do behave as 1:1 electrolytes,
but do not activate O2 under the same conditions. This is a particularly exciting result and
parsing out these differences is one of our primary goals in the future
work. In summary, our initial hypothesis that
fluorinated oxygen-donor ligands would serve as
medium field ligands has been proven spectroscopically and the reactivity studies of one
particular family, the Cu(I)-O2 system, shows significant promise
toward C-H activation with earth abundant metals and potentially catalytic
systems.