AmericanChemicalSociety.com
Reports: GB4 47864-GB4: Generation and Kinetic Studies of High-Valent Metal-Oxo Intermediates
Scientific and Technical Description of the Results
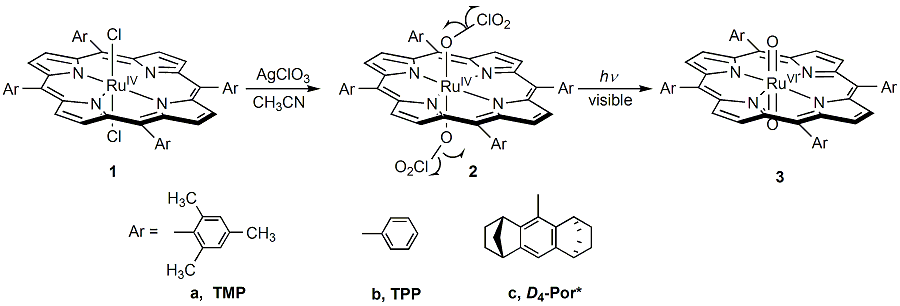
Photochemical generation of trans-dioxoruthenium(IV) porphyrin complexes
In literature, trans-dioxoruthenium(VI) porphyrin complexes have been received much attention, and developed as the well-characterized model system for heme-containing enzymes. Typically, trans-dioxoruthenium(VI) porphyrin complexes were prepared by oxidation of the corresponding ruthenium(II) carbonyl precursors with sacrificial oxidants such as m-CPBA or PhIO. Recently, we have successfully developed a new photochemical method that led to generation of the dioxoruthenium(IV) complexes bearing sterically hindered, unhindered and chiral porphyrin ligands (Scheme 1).
Scheme 1. Photochemical synthesis of trans-dioxoruthenium(VI)
porphyrins As shown in Scheme, the dichlororuthenium(IV) complexes (1) were first prepared. Exchange of the counterions
in 1 with Ag(ClO3) gave the
corresponding dichlorate salts 2. Irradiation
of dichlorate complexes 2 in anaerobic CH3CN
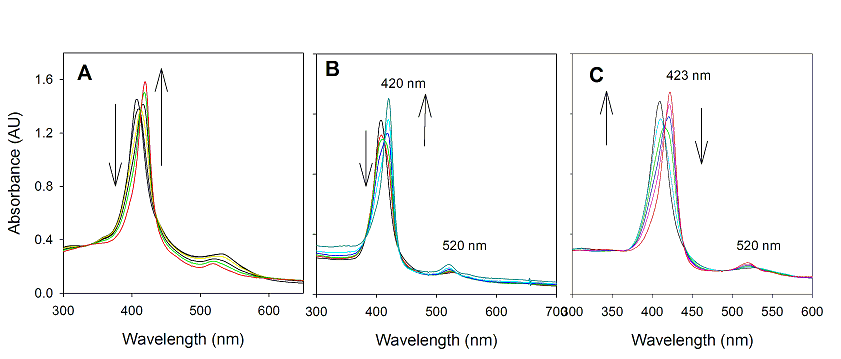
with visible light resulted in changes in absorption spectra with isosbestic points (Figure 1A). In Figure 1, 2a was decayed, and a new
species 3a was produced, displaying
a stronger Soret band at 420 nm and weaker Q band at
518 nm that is characteristic for RuVI(TMP)O2. The spectra signature of RuVI(TMP)O2
was further confirmed by 1H NMR and IR. Thus, the photolysis
reactions of the dichlorate complexes 2
undergo the homolytic cleavage of the O-Cl bond in the two chlorate counterions
simultaneously to produce neutral dioxoruthenium(VI)
species 3 via two one-electron photooxidation
pathways.
Figure 1 (A) Time–resolved UV-visible spectra of 2a (8 × 10-6 M) upon irradiation with visible light in
anaerobic CH3CN solution at 22 oC
over 50 min. (B) UV-visible spectral
change of 2b (8.0 × 10-6
M) upon irradiation over 45 min. (C)
Time-resolved
spectrum following irradiation of 2c
(1.0 × 10-5 M) over 60 min.
In a similar fashion, the sterically unhindered RuVI(TPP)O2
(3b) and chiral RuVI(D4-Por*)O2
(3c) were also generated (See Figure 1B and 1C). The use of other solvents such as CH2Cl2
gave the same results. The product degradation was observed when higher-energy
UV light (lmax = 350 nm)
was used instead of the visible light.
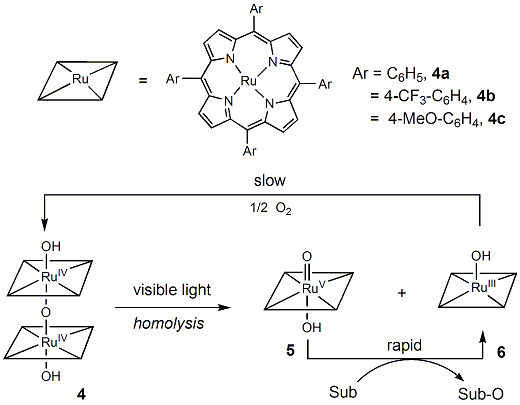
A series of ruthenium(IV)-m-oxo bisporphyrins
was evaluated in the aerobic oxidation of cis-cyclooctene (Table 1). After 24 hours of photolysis with
visible light (lmax = 420 nm), cis-cyclooctene oxide was
obtained as the only identifiable oxidation product (> 95% by
GC) with ca. 220 turnovers of catalyst 4a
(entry 1). The use of other solvents instead of CH3CN
resulted in reduced TONs (entries 2-4). Catalyst degradation was
a problem with higher-energy light, but the use of UV irradiation increased
catalytic activity (entry 5). The catalytic activity was enhanced by adding
small amounts of anthracene (entries 6 and 9). Quite
surprisingly, the axial ligand on the metal had a
significant effect, and the [RuIV( Table 1. Aerobic Photocatalytic Oxidation of cis-Cyclooctene
with Diruthenium(IV) m-Oxo Porphyrins a
Entry Catalyst Solvent T/day TONb,c 1 [RuIV(TPP)OH]2O CH3CN 1 220 4a 2 460 3 640 2 CHCl3 1 110 3 C6H6 1 140 4 THF 1 190 5d CH3CN 1 340 6e CH3CN 1 300 7 [RuIV(TPP)Cl]2O CH3CN 1 70 8 9e [RuIV(4-CF3-TPP)OH]2O 4b CH3CN CH3CN 1 1 250 340 10 [RuIV(4-MeOTPP)OH]2O 4c CH3CN 1 190 aThe reaction was
carried out with 0.5 µmol of catalyst
in 5 mL of solvent containing 4 mmol
of cis-cyclooctene.
Oxygen-saturated solutions were irradiated with visible light (lmax= 420 nm) or otherwise noted. b TON
represents the total number of moles of product produced per mole of catalyst.
All reactions were run three times, and the data reported are the averages. c The major product was cis-cyclooctene
oxide, detected in > 95% yield. d UV-visible light (lmax = 350 nm). e 5
mg of anthracene was
added. The photocatalytic
oxidations of a variety of organic substrates were examined in a similar way. Table 2 lists the oxidized products and
corresponding TONs using 4b as the photocatalyst. Activated hydrocarbons including triphenylmethane, diphenylmethane, ethylbenzene and xanthenes were oxidized to the
corresponding alcohols and/or ketones from
over-oxidation with total TONs ranging from 560 to 2900 (entries 3-6).
Noticeably, the oxidation of secondary benzylic
alcohols gave the highest catalytic activities (entries 7-8). Competitive
catalytic oxidation of ethylbenzene and ethlybenzene-d10 revealed a kinetic
isotope effect (KIE) of kH/kD =
4.8 ± 0.2 at 298 K. Table 2.
Turnover Numbers for
Alkenes and Benzylic C-H Oxidations Using 4b as the photocatalyst
a
Entry Substrate Product TONb 1 norbornene norbornene oxidec 200 2 cyclohexene 2-cyclohexenol 2-cyclohexenone cyclohexe oxide 160 350 30 3 triphenylmethane triphenylmethanol 1120 4 diphenylmethane diphenylmethanol benzophenone 820 140 5 ethylbenzene 1-phenylethanol acetophenone 380 180 6d xanthene 9-xanthone 2900 7 1-phenylethanol acetonphenone 3300 8 9-xanthenol 9-xanthone 3900 a
Typically with 0.25 µmol of 4b in CH3CN containing 4 mmol of
substrate and 5 mg anthracene. b Determined for a 24 h photolysis (lmax = 420 nm). c > 90% exo isomer. d One minor
product was detected by GC but not identified. Conclusions In summary, we report a new preparation of trans-dioxoruthenium(VI)
porphyrin complexes by an extremely easy
photochemical approach that bypassed the limitation observed in chemical
methods. We have also demonstrated
that the ruthenium(IV)-m-oxo bisporphyrins catalyzed efficient aerobic oxidation of
alkenes and activated hydrocarbons using visible light and atmospheric oxygen.
The observed photocatalytic oxidation is ascribed to
a photo-disproportionation mechanism to afford a
putative porphyrin-ruthenium(V)-oxo species as the active oxidant.