AmericanChemicalSociety.com
Reports: G6 47880-G6: Molecular Basis of Tetrahydrofuran-Induced Enclathration
The goal of this project is to understand the role of an additive, tetrahydrofuran (THF), in promoting the enclathration of methane. A molecular-scale understanding of this process requires understanding the physics of hydration and how additives change the solubility of the solute of interest (here methane). Towards this goal, we are developing tools and techniques to study the thermodynamics of hydration at the molecular scale. A key feature of our work is the ability to parse the excess chemical potential of the solute into (1) a chemical term, arising due to the interaction of the solute with water molecules within the defined coordination sphere (the inner-shell), (2) a packing contribution, accounting for forming a solute-free coordination sphere (the observation volume) in the solvent, and (3) a nonspecific, long range correction, accounting for the interaction of the ion with the solvent outside the coordination sphere. The theoretical developments here are also of broad interest in solution thermodynamics.
Ensemble reweighing technique for packing and chemistry: As noted in the earlier report, we developed a molecular aufbau approach to obtain the local chemical contribution. In this approach we populate the initially empty coordination shell with water molecules one at a time — in effect, we build the solution around the solute. For ions, this approach helped identify the number of water molecules that are sensitive to ion-type and quantified the range of ion-specific effects on the solvent. A key result was that low coordination states are most sensitive to ion-type and contribute most to the local chemical contribution. Our results are in qualitative agreement with spectroscopic studies of ions in water.
For strong solute-water interactions and/or for coordination spheres that are large (> 3.5 Å), it is difficult to observe the chemically interesting low coordination states: at thermal energies water does not spontaneously evacuate the coordination region around the solute. We have been testing an ensemble reweighing technique to efficiently map the low coordination states. In this approach we apply external weights that penalize the more probable coordination states and favor the low coordination ones. The final simulation statistics are then corrected for the applied weights.
Fig. 1 shows results (Merchant, Asthagiri; In preparation) for opening large, spherical cavities in water. For molecular solutes, it is desirable to have coordination spheres that are not spherical. As the schematic Fig. 2 shows, for a planar molecule such as THF, having a spherical cavity includes interactions that are not of direct interest to the local solute-water interaction. We have since developed an approach to study molecularly shaped cavities (Merchant, Dean, Asthagiri: 2010a, In preparation). The shaped-cavity is defined by overlapping spheres, the radii of which are determined from the proximal solute atom-water correlation function (Fig. 3). Fig. 4 shows the results for population distribution within a neopentane-sized cavity in water.
External molecular fields for hydration: In the molecular aufbau approach we assess the local contribution in the presence of the external solvent medium. We are investigating ways to describe the effect of the medium outside the coordination sphere by means of external fields. We obtain the field by an inverse sampling procedure. The water structure in the coordination sphere in the presence of the external medium is used as a reference. (All simulations are using empirical potentials.) For the same cluster, we perform simulations without the external medium but in the presence of a field. The field is adjusted such that the cluster distribution matches the reference (Fig. 4). For the hydration of methane, this approach is able to reproduce the local contribution (Merchant, Dean, Asthagiri; 2010b, In preparation). With this field, it should also be possible to study the solute-water cluster using quantum-chemical methods, an aspect that we will explore.
Ab initio hybrid Monte Carlo (AIHMC): In support of our broader goals to study hydration with chemical realism, we have developed an ab initio hybrid Monte Carlo method: after a defined number of molecular dynamics steps, configurations are chosen based on a Metropolis scheme. The forces are obtained by solving the electronic structure problem.
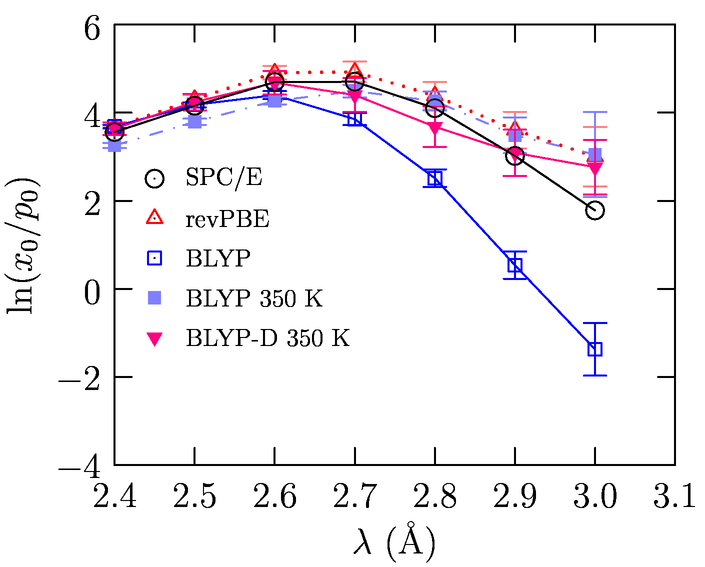
Using this scheme and long simulations, we studied water simulated with different electron density functionals. These studies show that dispersion-corrected functionals appear to reproduce the structure of the fluid and the balance of chemical and packing effects in hydration (Fig. 4; Weber et. al., JCP, 132, 204509).
We have also conducted even more extensive simulations with the BLYP-D density functional for two different system sizes. Using the quasi-chemical organization of the potential distribution theorem, we have computed the excess free energy of the liquid. With a separate calculation of the excess internal energy, we then obtained an estimate for the excess entropy. We find that although the free energy and the structure are well reproduced, the excess internal energy and entropy are in error relative to experiments. The manuscript on this has been reviewed favorably and we are preparing to submit the revised version (Weber and Asthagiri, 2010).
We plan to use the AIHMC method to probe the local water structure around THF and ethylene oxide (a simpler version of THF) to cross-check simulations with empirical parameters.
FIGURES
Fig. 1: Free energy of creating a large cavity in water. The
agreement with perturbation approach is excellent. The small
disagreement with scaled particle theory reflects system size effects.
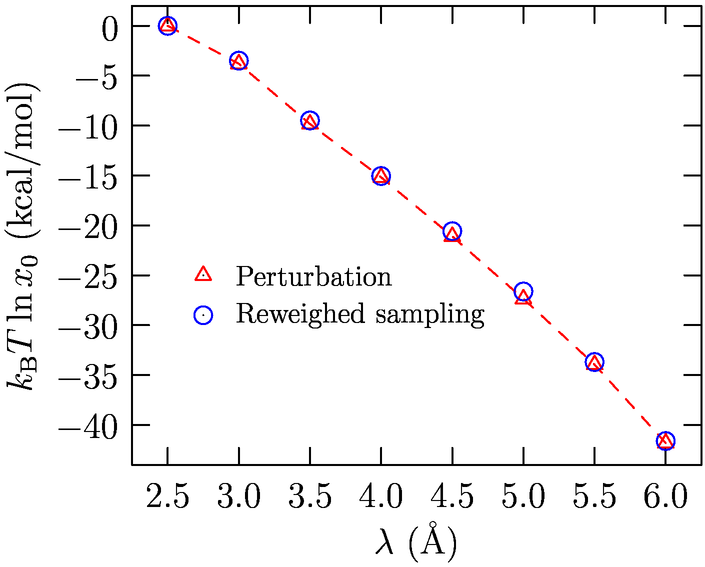
Fig. 2: Understanding water distribution in the proximal volume is more
insightful than the distribution in a spherical volume about molecular
center.
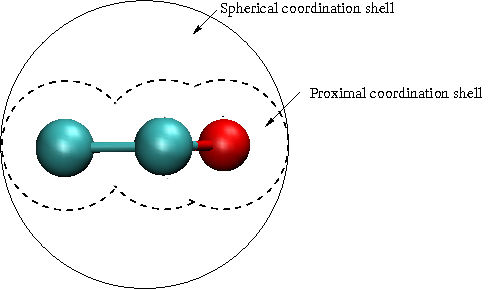
Fig. 3: Proximal methyl-water pair-correlation for neopentane compared
with methane-water pair correlation. The radius of atom representing
each methyl center is given by g(rprox)=1.0; rprox=3.35 A.
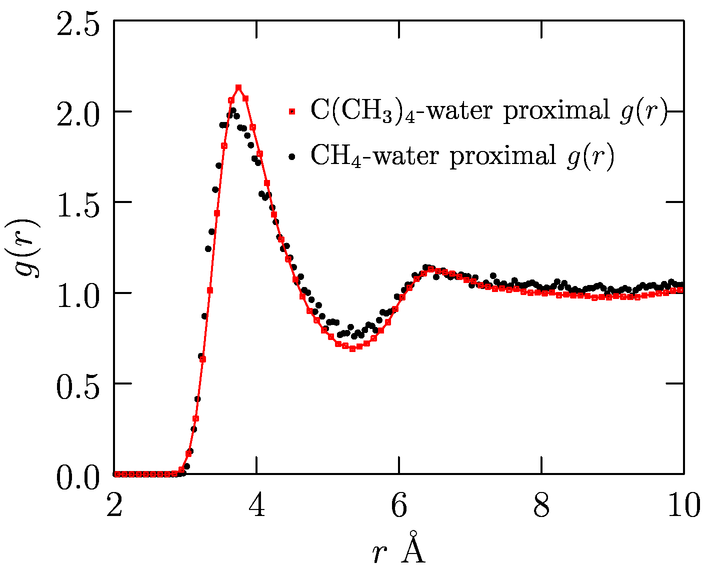
Fig. 4: Distribution of water molecules in a neopentane-shaped cavity, with radius for each methyl center obtained from Fig. 3.
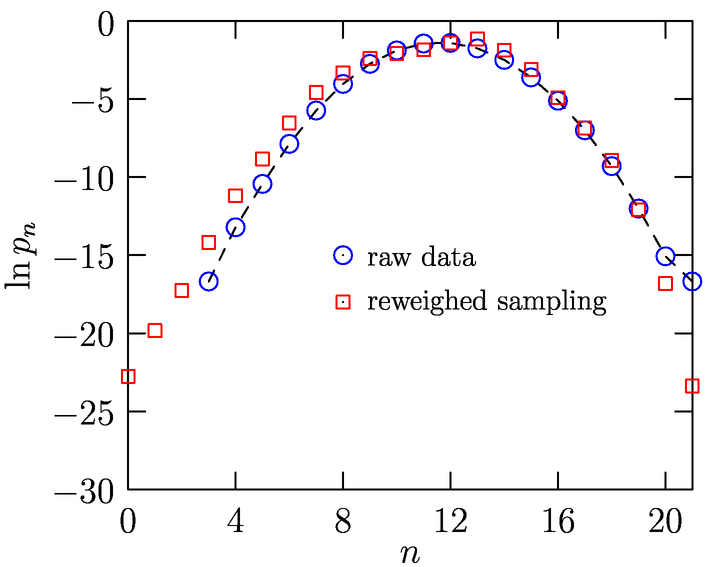
Fig. 5: Distribution of two water molecules in a cluster with and without field.
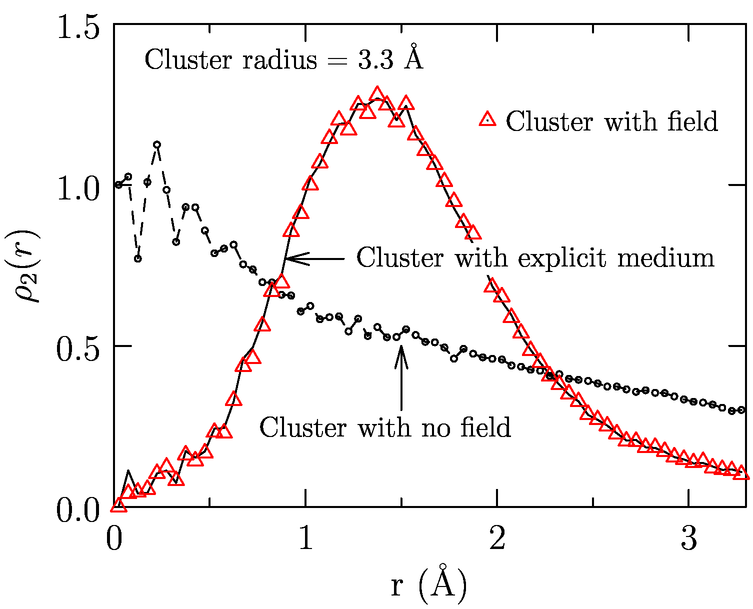
Fig. 6: Balance of local chemistry and packing in water simulated with various electron density functionals.