AmericanChemicalSociety.com
Reports: DNI1 49707-DNI1: Low-Valent Iron-Catalyzed Transformations of Unsaturated Hydrocarbons
Since work by Kharash in 1941, development of iron catalysts for synthetic chemistry has shown that iron is useful in carbon–heteroatom and carbon–carbon bond forming reactions. Our proposal outlined two major aims: (1) to develop iron-catalyzed transformations to access building blocks from commodity chemicals such as dienes, which can be directly derived from petroleum and (2) to improve understanding of carbonyl-free low-valent iron catalysis. During the last ACS-PRF granting year, our laboratory reported the development of iminopyridine-iron catalysts for the selective 1,4-addition of alpha-olefins to 1,3-dienes and the chemo-, regio-, and stereoselective 1,4-hydroboration of 1,3-dienes.
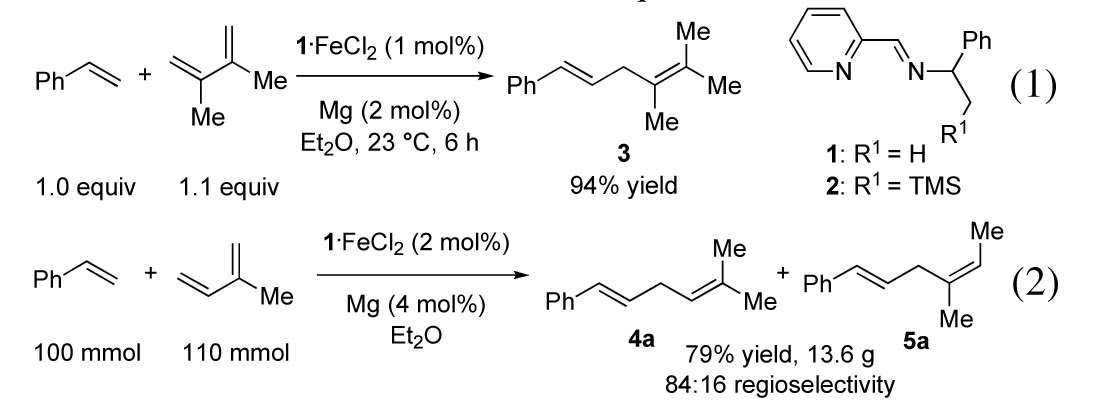
We have developed a functional group tolerant, stereo- and regioselective iron-catalyzed 1,4-addition reaction of terminal olefins to 1,3-dienes. The linear 1,4-diene products cannot be obtained with other iron catalysts or by any other hydrovinylation reaction reported to date.
The readily available iron(II) complex 1-FeCl2 can be reduced in situ with activated magnesium metal to catalyze carbon–carbon bond formation to afford 1,4-diene 3 (eq 1).
While the reaction shown in eq 2
afforded 1,4-addition product in 79% yield, two isomeric products were obtained
due to bond formation at either of the two different olefin termini of
isoprene. Ligand 1 produced prenylation product 4a selectively; the introduction of a
prenyl group upon 1,4-addition to isoprene is synthetically useful, but
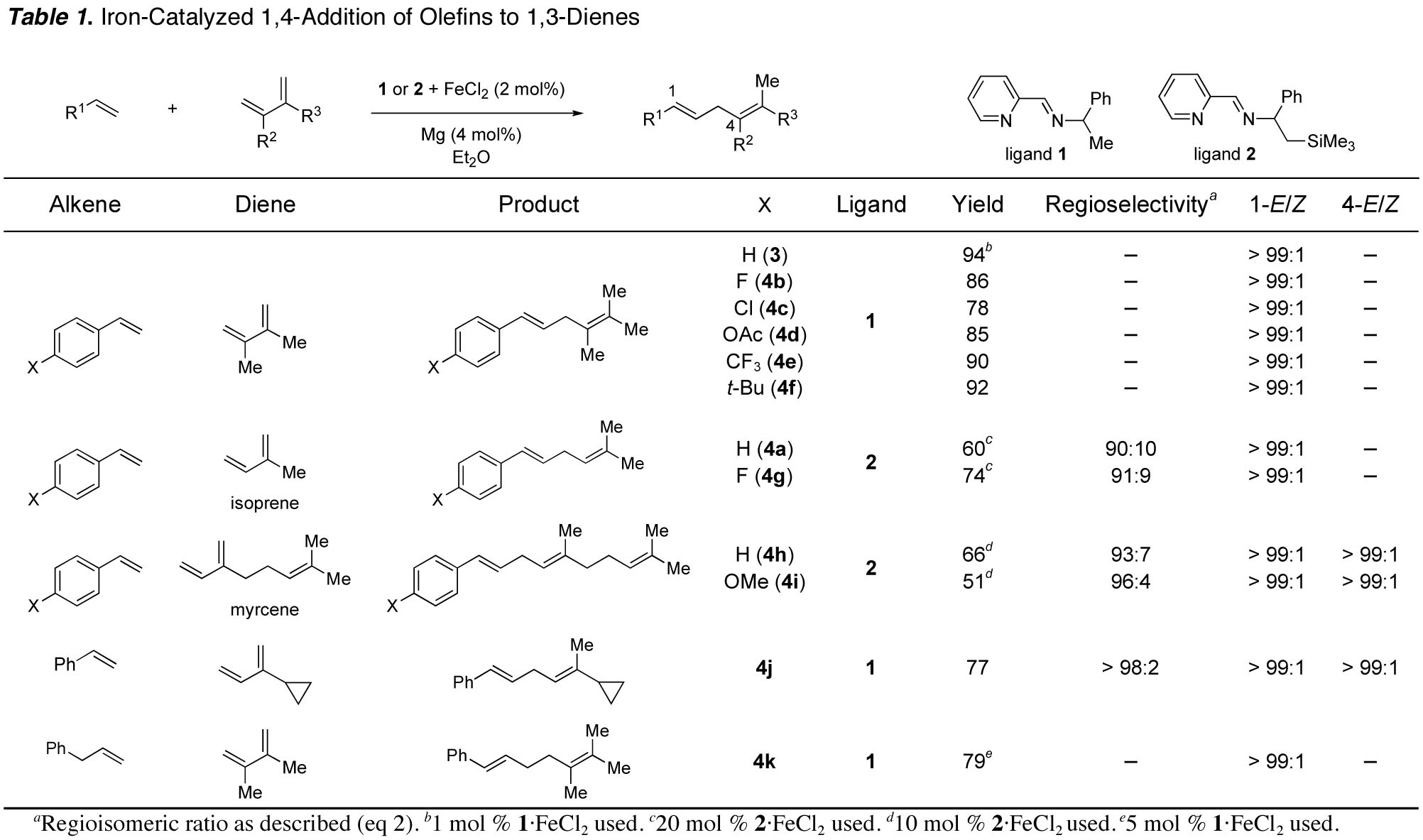
control over regioselectivity is challenging. To increase the regioselectivity, we evaluated different
iminopyridine ligands and found a strong dependence of the regioselectivity on
the ligand. The highest ratio of
10:1 favoring linear 1,4-diene was observed with the trimethylsilyl-substituted
ligand 2 (Table
1).
A
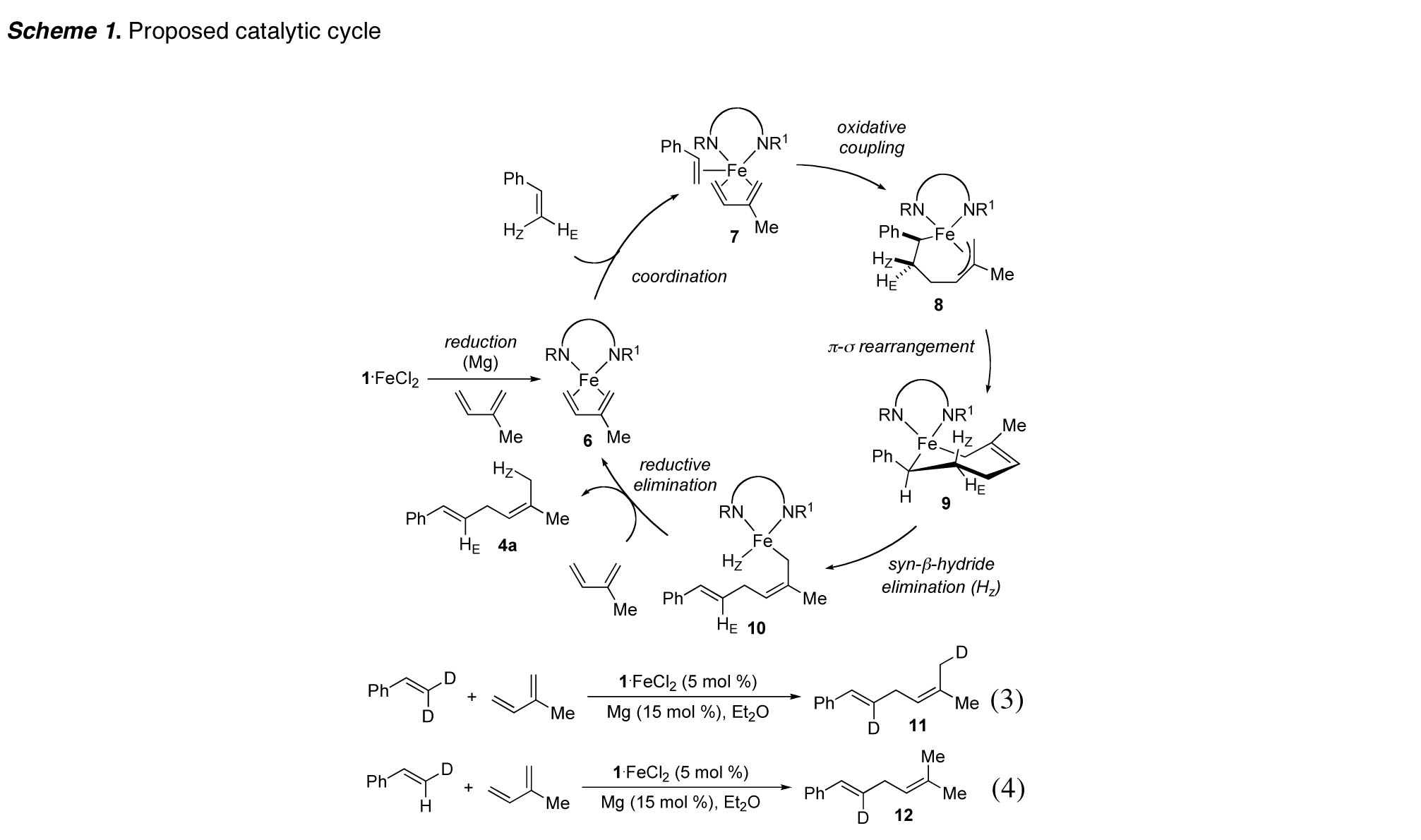
mechanistic hypothesis consistent with our experimental observations is detailed in Scheme 1. We propose formation of complex 8 after reduction of the Fe(II)
complex 1-FeCl2
and oxidative coupling with styrene.
At this time, we cannot yet rationalize the influence of the catalyst on
the regioselectivity, which may be determined during oxidative coupling. A pi-sigma rearrangement to form the
ferracycle 9, in
which the phenyl substituent is pseudoequatorial, positions only one
beta-hydride (Hz) syn to iron.
Hence, beta-hydride elimination can only afford a single double bond
isomer and delivers the 1,2-disubstituted E double bond as part of the iron(II)
alkyl hydride 10. Assuming that subsequent reductive
elimination proceeds without sigma-pi-sigma rearrangement of 10, the new trisubstituted double bond
is formed with E geometry
stereospecifically. To
substantiate the assumption that stereospecific reductive elimination from 10 is faster than isomerization, we
designed two deuterium-labeling experiments. The deuterium atoms of beta,beta-dideuterostyrene and (E)-beta-deuterostyrene, following
addition to isoprene, were observed exclusively at the positions indicated in 11 and 12, respectively, consistent with our
mechanistic hypothesis (eqs 3 and 4).
We also reported carbon–boron
bond formation by 1,4-hydroboration of 1,3-dienes using related
iminopyridine-iron catalysts. The allylborane products are formed chemo-,
regio- and stereoselectively with (E)-double bond geometry exclusively and are challenging to
access selectively with conventional chemistry. To our knowledge, there are no other examples of
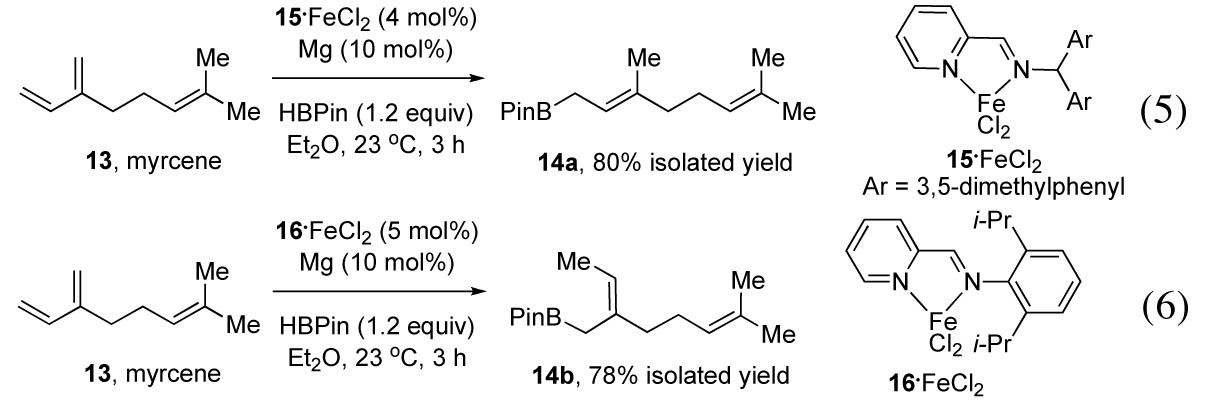
iron-catalyzed hydroboration reactions of olefinic substrates. Ligand optimization revealed that
the substituent on the imine nitrogen modulates the 1,4-regioselectivity to
favor either the branched or the linear isomer (eqs 5 and 6). 1,4-Addition of pinacolborane to
myrcene (13)
catalyzed by 15-FeCl2
produced geranylpinacolborane (14a) selectively (eq 5).
When catalyst 16-FeCl2 was used, the regioselectivity inverted to afford branched
allylborane 14b
as the major product (eq 6).
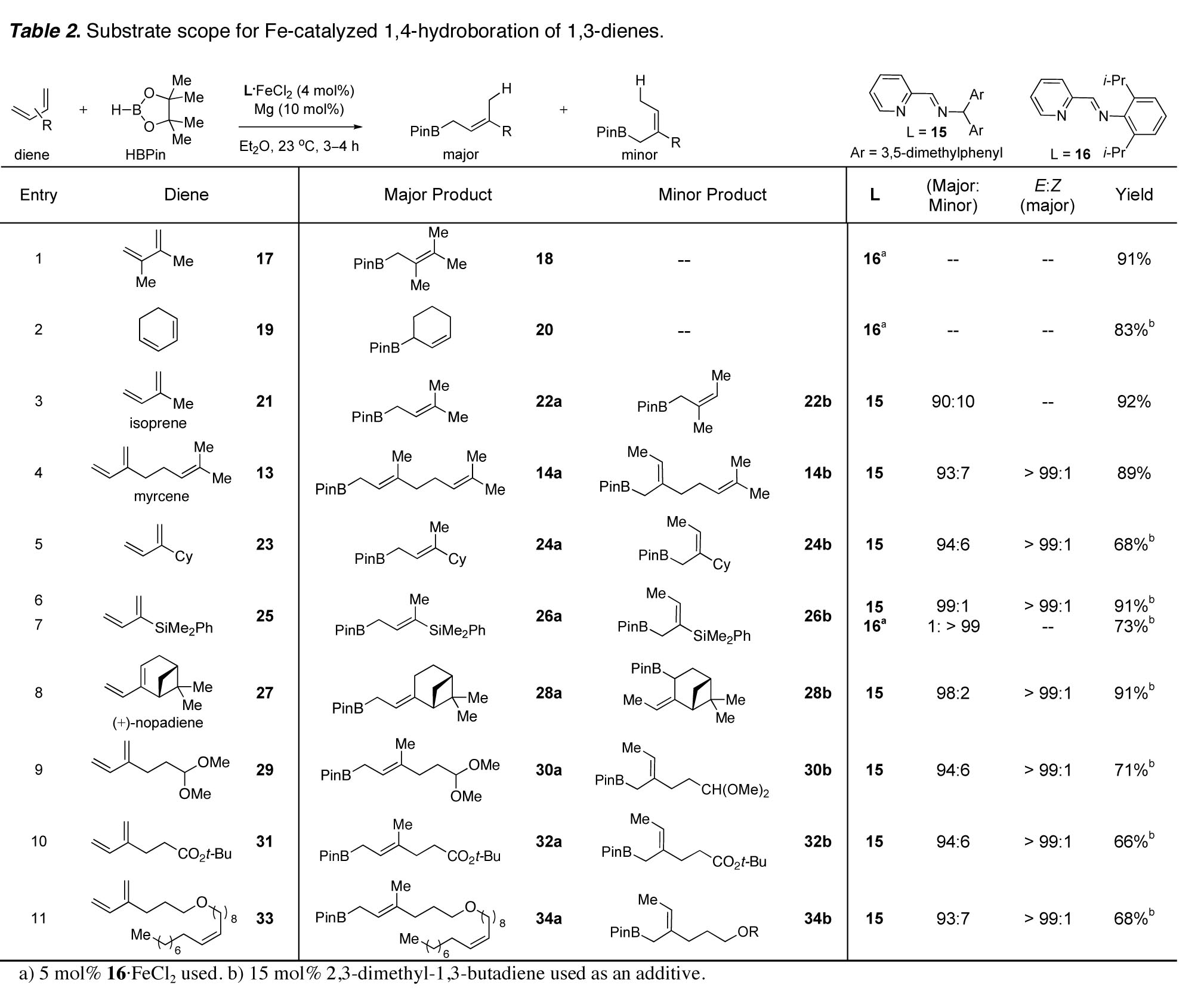
As shown in Table 2, the
regioselectivity for 1,4-hydroboration of 2-substituted dienes increased as the
size of the 2-substituent increased. The Fe-catalyzed hydroboration can be
performed in the presence of electrophilic functionality (e.g. ester 31) that is incompatible with the basic
conditions of traditional allylborane syntheses. Notably, hydroboration is chemoselective, and 1,4-addition
to 1,3-dienes proceeds in the presence of isolated olefins (33). The major linear isomers were formed with (E)-double bond geometry; no Z-olefins were detected for any
example. (E)-Trisubstituted double bonds are
otherwise challenging to synthesize stereoselectively, especially when the
substituents are similar in size.
In summary, we have reported regio-
and stereoselective iron-catalyzed 1,4-addition reactions to 1,3-dienes to
afford linear 1,4-dienes and (E)-gamma-disubstituted allylboranes. The iron-catalyzed reactions provide
access versatile building blocks that cannot be readily prepared by traditional
syntheses or other known transition metal-catalyzed reactions. Additionally, the hydroboration
reaction demonstrates previously unknown reactivity for iron. Ongoing investigations in our
laboratory include mechanistic studies to understand the role of the
iminopyridine ligand in catalysis as well as application of the catalysts
described herein to the development of new reactions.