AmericanChemicalSociety.com
Reports: DNI1 49965-DNI1: Catalytic Scaffolding Ligands: An Efficient Directing Group Strategy
With the support of the PRF over the past year, we have applied our scaffolding ligands to challenging problems in hydroformylation. The majority of our efforts have focused on the development of a method for the formation of quaternary carbons through hydroformylation and the synthesis of an enantioenriched variant of ligand 1.
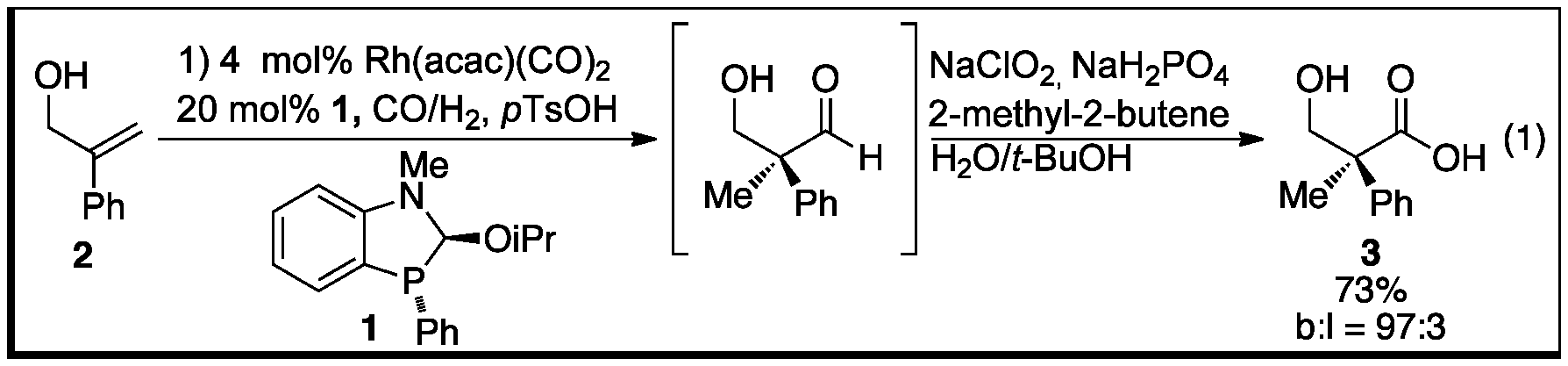
The synthesis of quaternary carbon centers has been a long-standing problem in the area of hydroformylation. The formation of quaternary centers via hydroformylation is so unfavourable that in 1948 Keulemans stated that, "addition of the formyl group to a tertiary C atom does not occur, so that no quaternary C atoms are formed" (Keulemans' rule). Inspired by previous work on phosphorous-directed hydroformylation performed in the Leighton group, we applied our strategy towards the hydroformylation of 1,1-disubstituted olefins towards the synthesis of quaternary carbon centers. Our initial attempts at hydroformylation of 2 showed significant formation of an aldehyde product (~50% 1H NMR yield); however attempts to isolate the product led to poor isolated yields. In order to improve the isolated yield, a Pinnick oxidation was performed directly after hydroformylation affording the more stable carboxylic acid product. Under these modified conditions acid 3 was isolated in 73% yield with excellent branch selectivity (b:l = 97:3, eq 1). The improved isolated yield over the 1H NMR yield of the aldehyde product was attributed to reversible dimerization of the b-hydroxy-aldehyde. Under the oxidation conditions the dimer can revert to the aldehyde, subsequently being oxidized to 3. With the optimal conditions in hand, we evaluated the substrate scope and found that a range of functional groups (e.g. ester, nitrile, bromo, chloro) and heterocyclic rings (e.g. pyridyl, fural) provide good yields and excellent regioselectivities for the branched aldehyde products.
Concurrently, we developed an
enantioenriched scaffolding ligand.
The first attempt at obtaining an enantioenriched ligand was through
resolution of racemic 1. An enantiopure alcohol was exchanged
onto 1 generating two diastereomeric
ligands. The exchange reaction was
successful; however, a 3:1 mixture of diastereomers was observed rather than
the expected 1:1 mixture. These results
are consistent with both the phosphorous and carbon stereocenters epimerizing
under the reaction conditions. To
circumvent this problem, we designed ligand (MeO)-4a, which contains an additional stereocenter that is readily
incorporated into the ligand synthesis via an enantioselective asymmetric
hydrogenation of 2-isopropylquinoline.
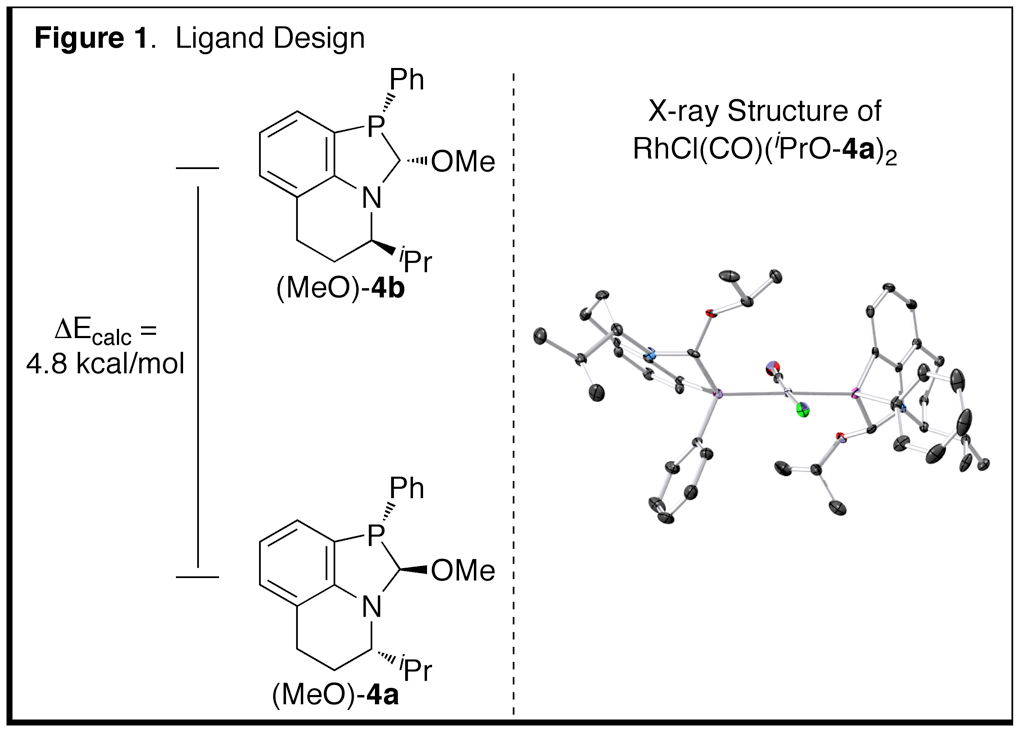
Computational studies suggested that (MeO)-4a is 4.8 kcal/mol more stable than (MeO)-4b (Figure 1). The
greater stability of 4a is
attributed to minimization of a syn-pentane-like interaction between the
isopropyl group and the C-O bond in the anti-anti
conformation. Consistent with this
hypothesis, (MeO)-4 was synthesized
as a kinetic mixture of 4 diastereomers that is converted to a single
diastereomer when equilibrated with isopropanol and 4 molecular sieves. X-ray crystallography confirmed the
relative and absolute stereochemistry of the ligand, which matched the
computational results (Figure 1).
With 4a in hand we applied the ligand in the
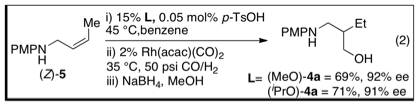
asymmetric hydroformylation of allylic amines towards the synthesis of b-amino-aldehydes. Hydroformylation of (Z)-5
with either (iPrO)-4a or
(MeO)-4a yields the desired b-amino
alcohol in good yield and up to 92% ee (eq 2). The optimized conditions are very mild (35 °C and
50 psi CO/H2), demonstrating the power of directing groups to
accelerate reactions. (Z)-olefins were generally found to yield
the product in high enantioselectivities between 86-94% ee. (E)-olefins,
as well as the terminal substrate, afford the products with promising levels of
enantioselectivity (72-80% ee). We
are currently investigating other ligand derivatives in order to improve the
selectivities towards more practical levels.
In
conclusion this past year we have addressed a significant problem in
hydroformylation, the formation of quaternary carbons. Furthermore, we have developed an
enantioselective catalyst system for the asymmetric hydroformylation of allylic
amines. We are continuing to
explore this novel ligand class in regio-, diastereo- and enantioselective
hydroformylation reactions.