AmericanChemicalSociety.com
Reports: UR1 49493-UR1: Development of Enantioselective Nitrogen Arylation Reactions
The development of new reaction methodologies capable of enantioselectively forming chiral products from achiral starting materials is an area of intense research. When the selectivity in such reactions is induced from only a very small amount of chiral catalyst, the methodology can be a cost-effective way to form very valuable chiral products from relatively inexpensive starting materials. With this goal in mind, we have been exploring the utility of the Buchwald-Hartwig reaction as an enantioselective reaction.
In general, a Buchwald-Hartwig reaction is a palladium-catalyzed cross-coupling reaction between a nitrogen nucleophile, and an aryl halide, forming an aromatic C-N bond. While the literature contains hundreds references to this important reaction, fewer than ten studies have explored the possibility of using a Buchwald-Hartwig reaction to induce asymmetry in a product, and most of these achieved only modest selectivities.
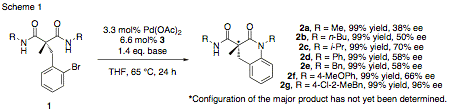
Our investigation began with the synthesis of symmetrical malonamides 1 (Scheme 1), in which the two identical amide groups are enantiotopic. By subjecting 1 to intramolecular cross-coupling, six-membered ring 2 is formed, and the symmetry of the molecule is broken, rendering the alpha carbon into a chiral center. A screen of catalyst systems incorporating chiral phosphines as ligands was undertaken, and it was found that (R)-MOP (4, in Scheme 2) was most capable of inducing the highest enantioselectivity. Under optimized conditions, for substrates 1 with R groups varying in steric bulk from methyl up to 4-chloro-2-methylbenzyl, products 2 were isolated in excellent yields. Interestingly, the relatively sterically unencumbered methyl-substituted 2a was formed in only 38% ee, but increasing steric bulk improved selectivity, with the best result being the bulkier 2g, which was isolated in 96% enantiomeric excess.
The lower observed enantioselectivity for less sterically
bulky substrates has prompted us to explore whether it may be possible to
improve selectivity by tuning the steric bulk of the catalyst. Toward this end, we have developed a
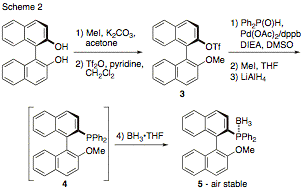
modification to the literature synthesis of 4, in which the phosphine functionality is installed at a later stage,
which should permit us to introduce new phosphine substituents with greater
steric bulk than phenyls, all from a late-stage common intermediate 3. In
addition to the new synthetic route, we have found that it is convenient to
isolate and store 4 as the borane
adduct 5. This complex can be handled (including
chromatography, storage, dispensing, etc.) without the need to take precautions
to exclude air. We are currently
investigating whether 5 can be
used directly in cross-coupling reactions as an in situ source of
4.
Current ongoing projects supported by this grant include: 1)
Applying our new route to the synthesis of new phosphine ligands related to 4, and exploring their use in the established
desymmetrization reaction; 2) Expanding the substrate scope of enantioselective
intramolecular Buchwald-Hartwig reactions, to include new ring sizes and new
heterocycles; and 3) Extending the concept of desymmetrizing Buchwald-Hartwig
reactions to intermolecular reactions.