AmericanChemicalSociety.com
Reports: G10 47332-G10: Synthesis, Structural Characterization and Property Optimization of Novel Antimonides and Bismuthides – Promising Materials for Thermal-to-Electric Energy Conversion
Introduction
In the last decade, there have been numerous reports on intermetallic compounds of the pnicogen elements with the alkali or alkaline-earth metals and many of the 3d-metals. More recently, the interest in such materials has further increased, largely due to the unexpected discovery of superconductivity in potassium-doped (Ba1-xKx)Fe2As2, RbFe2As2 and CsFe2As2, LiFeAs, LiCu2P2, etc. The appeal of many antimonides for use in thermoelectrics, such as (Ca1-xYbx)Zn2Sb2, Yb14MnSb11, SrZn2Sb2, EuZn2Sb2, BaZn2Sb2, among others, has also contributed to the growing attention in this sub-field of chemistry.
The past research efforts in our group have been closely aligned with research on the crystal and electronic structures of new Zintl phases with complex structures, both from a fundamental and practical standpoint. Inspired by their promise for solid-state energy conversion, we have already reported on more than a dozen novel ternary pnictides with the d5- and d10-metals, whose bonding characteristics could be advantageous in the efforts to optimize the thermoelectric efficiency. Most of the previous work has been focused on A-Cd-Pn phases, where A = Ca, Sr, Ba, Eu, Yb; and Pn = As, Sb, Bi. Extending our research into the zinc-pnictides, we first undertook systematic syntheses in the Ba-Zn-Pn ternary systems, which seem sporadically and insufficiently mapped out. So far, only the following four phases have been synthesized and structurally characterized—BaZn2P2 (ThCr2Si2 type), the dimorphic BaZn2As2 (BaCu2S2 and ThCr2Si2 type), BaZn2Sb2 (BaCu2S2 type), BaZnSb2 and BaZnBi2 (ZrAl3 type). Herein we report the synthesis and the crystal structures of three new Zintl phases Ba2ZnAs2, Ba2ZnSb2 and Ba2ZnBi2, effectively doubling the number of ternary compounds in each of the corresponding phase diagrams.
Results
Because the title compounds are isotypic and isoelectronic, for the sake of simplicity, the focus of the structural discussions will be primarily on one of them, Ba2ZnSb2. In the following two paragraphs, we will briefly describe the important characteristics of its structure. We will also depict some useful structural relationships and examine in greater detail the chemical bonding.
Structure. Ba2ZnSb2 crystallizes in the centrosymmetric space group Ibam (Figure 1) with three crystallographically unique atoms in the asymmetric unit, all in special positions.
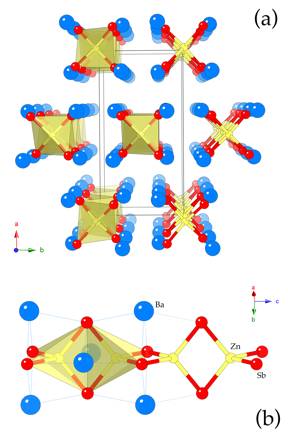
Figure 1. (a) Combined ball-and-stick and polyhedral view of the
Ba2ZnSb2 structure, projected down the c-axis. The representation emphasizes the isolated ![]() [ZnSb2]
polyanionic chains, "solvated" by the cations. The orthorhombic unit cell is
outlined. (b) A close-up view of a
segment of the
[ZnSb2]
polyanionic chains, "solvated" by the cations. The orthorhombic unit cell is
outlined. (b) A close-up view of a
segment of the ![]() [ZnSb2]
chains, depicting the edge-shared ZnSb4 tetrahedra. The Ba cations,
which cap the triangular faces in m3-fashion
are also shown. Color code: Zn – yellow; Sb – red; Ba –
blue, respectively.
[ZnSb2]
chains, depicting the edge-shared ZnSb4 tetrahedra. The Ba cations,
which cap the triangular faces in m3-fashion
are also shown. Color code: Zn – yellow; Sb – red; Ba –
blue, respectively.
Formally, the structure belongs to the known K2SiP2 type (Pearson's code oI20), where Ba takes the K site, and the Zn atoms are at the Si site. Ba2ZnSb2 is also isostructural and isoelectronic to the ternary chalcogenides A2MnQ2 and A2CoQ2 (A = K, Rb, Cs; Q = S, Se, Te). Noteworthy, all previously known compounds with this structure contain alkali metals as cations, while Ba2ZnPn2 (Pn = As, Sb, Bi) appear to be the first phases formed with alkaline-earth metals cations; they are also the first antimonides and bismuthides in this series.
Following the differences in
electronegativity between the constituting elements, the Ba2ZnSb2 structure (Figure 1a) can be "broken down" to Ba2+
cations and isolated polyanionic chains, ![]() [ZnSb4/2]4–,
running parallel to the c-axis. They
are made of edge-shared ZnSb4 tetrahedra
(Figure 1b), and are isosteric to the
[ZnSb4/2]4–,
running parallel to the c-axis. They
are made of edge-shared ZnSb4 tetrahedra
(Figure 1b), and are isosteric to the ![]() [SiS4/2] chains in the binary SiS2. The chains
are indeed well-separated from one another with closest contacts between
adjacent Sb-vertices on the order of 5.3 (center-to-center separation is ca.
7.9 ). Within the ZnSb4 tetrahedra, Zn–Sb
interactions appear to be typical 2-center 2-electron bonds with distances that
measure 2.7744(3) . These values compare well with those reported for other
intermetallic compounds with similar bonding patterns: dZn-Sb = 2.675-2.924 in Ca9Zn4.5Sb9,
dZn-Sb = 2.725
in Yb14ZnSb11, dZn-Sb = 2.660-2.810 in BaZn2Sb2,
dZn-Sb = 2.722
in NaZnSb, dZn-Sb
= 2.710-2.727 in REZn1–xSb2 (RE = La-Tb), etc. The tetrahedral angles deviate
slightly from the ideal 109.5°, but nothing out of the ordinary. Similar
comparisons can be made between the Zn–As (Zn–Bi) distances and angles
in Ba2ZnAs2 (Ba2ZnBi2) with the
metrics of other published structures.
[SiS4/2] chains in the binary SiS2. The chains
are indeed well-separated from one another with closest contacts between
adjacent Sb-vertices on the order of 5.3 (center-to-center separation is ca.
7.9 ). Within the ZnSb4 tetrahedra, Zn–Sb
interactions appear to be typical 2-center 2-electron bonds with distances that
measure 2.7744(3) . These values compare well with those reported for other
intermetallic compounds with similar bonding patterns: dZn-Sb = 2.675-2.924 in Ca9Zn4.5Sb9,
dZn-Sb = 2.725
in Yb14ZnSb11, dZn-Sb = 2.660-2.810 in BaZn2Sb2,
dZn-Sb = 2.722
in NaZnSb, dZn-Sb
= 2.710-2.727 in REZn1–xSb2 (RE = La-Tb), etc. The tetrahedral angles deviate
slightly from the ideal 109.5°, but nothing out of the ordinary. Similar
comparisons can be made between the Zn–As (Zn–Bi) distances and angles
in Ba2ZnAs2 (Ba2ZnBi2) with the
metrics of other published structures.
Ba-coordination is distorted octahedral with "normal" Ba–Sb distances. Four Ba2+ cations are positioned around each ZnSb4 tetrahedron in such a way (Figure 1b) that each of its faces is capped. The "solvation" of the anions by cations has also been discussed by Corbett on the example of K2SiAs2. Such an arrangement of the cations and anions results in Ba–Zn contacts, which are shorter than the sum of the Ba and Zn metallic radii (rBa = 2.215 ; rZn = 1.339 ), and shorter than the observed Ba–Zn distances in BaZn5, BaZn13, and all known BaZnPn2 and BaZn2Pn2 compounds. Although the above might be suggestive of a certain covalency of the Ba–Zn interactions, the electronic structure calculations discussed later (vide infra), indicate very weak Ba–Zn bonding.
The last point of relevance to the structural description
we touch upon concerns the Zn–Zn distances. Inspection of the structures
of the ternary pnictides BaZn2Pn2
and BaZnPn2 shows that
they feature anionic networks made of edge-sharing or combinations of edge- and
corner-sharing of ZnPn4
tetrahedra. As a result, Zn–Zn distances in these compounds are
relatively shorter than those in Ba2ZnPn2. For comparison, Zn–Zn contacts
in BaZn2Sb2 measure 3.241 , while in Ba2ZnSb2
dZn–Zn = 3.480
, respectively. This elongation can be related to the reduced dimensionality
of the anionic sub-structure in Ba2ZnPn2, compared to BaZn2Pn2, and the unique linear geometry of the ![]() [ZnSb4/2]4–
chains. Therefore, the contribution of the Zn–Zn interactions to the
overall bonding is expected to be negligible, and the structure can be viewed
as containing no close homoatomic bonds. Consequently, applying the simple
valence rules, Ba2ZnSb2 can be rationalized as (Ba2+)2(Zn2+)(Sb3–)2
or alternatively, as (Ba2+)2[(4b-Zn2–)(2b-Sb1–)2], i.e., a typical
Zintl compound. Electronic structure calculations presented below confirm the
description based on the classic Zintl formalism.
[ZnSb4/2]4–
chains. Therefore, the contribution of the Zn–Zn interactions to the
overall bonding is expected to be negligible, and the structure can be viewed
as containing no close homoatomic bonds. Consequently, applying the simple
valence rules, Ba2ZnSb2 can be rationalized as (Ba2+)2(Zn2+)(Sb3–)2
or alternatively, as (Ba2+)2[(4b-Zn2–)(2b-Sb1–)2], i.e., a typical
Zintl compound. Electronic structure calculations presented below confirm the
description based on the classic Zintl formalism.
