
Back to Table of Contents
42390-GB6
Simulating Linear and Nonlinear Optical Spectra in Condensed Phase Systems with a Mixed Molecular Dynamics and Quantum Mechanical Method
Brent P. Krueger, Hope College
Introduction Fluorescence-detected
resonance energy transfer (FRET) experiments are widely utilized to examine the
structure and structural dynamics of polymer and biopolymer systems. With a
variety of modern fluorescence spectroscopies that can be coupled to
microscopies, as well as the relative ease with which (two) fluorescence probe
molecules can be incorporated into polymers, FRET has become relatively easy to
employ in a qualitative fashion and has experienced a rebirth in the past decade
or so. However, quantitative analysis is hampered by the uncertainty
surrounding a number of approximations that must be made. Most famous of these
is the 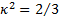 approximation, which requires that
both probes isotropically sample all possible orientations. Several others
also lead to difficulty, including the assumption of no correlation between the
probe-probe orientation factor (
approximation, which requires that
both probes isotropically sample all possible orientations. Several others
also lead to difficulty, including the assumption of no correlation between the
probe-probe orientation factor (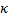 ) and the probe-probe distance, which
we recently demonstrated (VanBeek et al., 2007, Biophys. J., 92,
4168–4178).
) and the probe-probe distance, which
we recently demonstrated (VanBeek et al., 2007, Biophys. J., 92,
4168–4178). The two approximations
mentioned above are associated with the relative motions of the probe
molecules. Thus, if one could estimate the structural dynamics of the two
fluorescent probes these difficult approximations would not be needed and the
quantitative ability of FRET experiments would be improved. We have spent much
of the past year beginning molecular dynamics (MD) calculations that will be
used to simulate the structural dynamics. In addition, we have conducted
quantum mechanical (QM) calculations that will explore the breakdown of another
key FRET approximation—the ideal dipole approximation. Finally, we also began
experimental work, learning sample preparation and technical details and
collecting our own data using single-molecule fluorescence techniques. The
computer simulations will mesh with fluorescence data to provide an estimate of
the fluorescent probe motion, obviating the need for some approximations, and
therefore improving the quantitative capability of FRET experiments.Progress
The short-term goal for
the past year (year 3 of the grant) was to use calculations to help identify
what portion of the experimentally observed structural dynamics are due to the
biopolymer of interest, due to the probe molecules, and due to the linkers that
connect the probes to the biopolymer. We are focusing on several small RNA
constructs that are all derived from the hairpin ribozyme (HR)—a small
catalytic RNA molecule that exhibits a large structural change during
activation. The HR has been widely studied using ensemble and single-molecule
FRET experiments. However, it is not known how much the probes themselves (and
the linking moieties) contribute to the total system dynamics observed
experimentally.
To examine the structural
dynamics of the HR constructs we are using molecular dynamics (MD)
simulations. The force fields used in biomolecular MD simulation packages do
not have parameters for the fluorescent dyes of interest. So, this research
began by developing MD parameters for the fluorescent molecules and the
linkages that connect them to the nucleic acid constructs. This research was
performed by David Paul, the SUMR student supported by this grant. David
completed parameterization of several probe molecules as well as generic
linkage moieties for connecting any of the probes to DNA or RNA. These results
allowed David to construct simulation systems matching those that we are using
in the single-molecule experiments. Though MD simulations are
not yet complete, David's experience in performing parameterization has been
published as an “advanced tutorial” on the AMBER website. AMBER is a MD
package that is widely used by academics and industry for biomolecular
simulations, receiving 90-100 citations per year. Our tutorial is part of a
suite of tutorials that assist both novice and experienced AMBER users and
receive an average of over 50,000 hits per day. In addition to the
MD work, we have also used QM calculations to examine the breakdown of the
ideal dipole approximation (IDA). Conducted in collaboration with Dr.
Benedetta Mennucci's group at the University of Pisa, these calculations have
involved undergraduate Lydia Hartsell. Dr. Mennucci's group has developed a QM
description of the interaction between molecules that drives the energy
transfer necessary for FRET. This development, based on my previous work with
Dr. Graham Fleming and Dr. Greg Scholes, has allowed us to determine an
accurate value for the interaction at any distance, and therefore to quantify
breakdown of the IDA when the two fluorescent probes are close to each other.
A manuscript describing the result is currently preparation and will likely be
submitted in October.Summary
Over the three years of
this award my group has completed investigation of a hybrid MD-QM method for
examining solvation dynamics, has quantified the breakdown of the IDA, has
completed parameterization of several fluorescent probe molecules, has begun MD
simulation of RNA constructs for FRET experiments, and has begun single-molecule
FRET experiments of these same systems. This work has resulted in two
publications, with one more near submission, and an AMBER tutorial.
Back to top
