45130-G3
Redox Coupling for Multielectron Small-Molecule Activation
Our approach to this problem utilizes redox-active ligands to store
and deliver charge to later 3d metal centers for multielectron reactions with
small molecules. In this way, four- and five-coordinate manganese, iron and
cobalt centers act as surrogates for platinum group metal catalysts, particularly
those that rely on oxidative-addition and reductive-elimination steps in
catalytic cycles for small-molecule activation and functionalization. Progress Report. Chart 1 summarizes the redox-active ortho-catecholate (cat) and ortho-arylamidophenolate (apAr)
ligands used in this work. These ligands were selected because their frontier
orbitals are close in energy to the manganese, iron and cobalt 3d orbitals, and
modification of the ligand can be used to tune steric and electronic properties.
This approach has been fruitful. During the grant period we have discovered new
stoichiometric and catalytic ligand-mediated multielectron reactions at coordinatively
unsaturated first-row transition metals.
Cross coupling catalysis. We have prepared and
characterized a series of square planar cobalt complexes with two o-arylaminophenolate (apAr)
(Ar = Ph, 2,6-iPr2C6H3, 3,5-Cl2C6H3)
ligands. Addition of 1.0 equiv Cp*2Co or Na to blue CoIII(apAr)(isqAr)
species cleanly generates air-sensitive violet [CoIII(apAr)2]–
products (Figure 1).
The
S = 1 [CoIII(apAr)2]– complexes are
strong nucleophiles. Reaction of 1.0 equiv of
2,3,4,5,6,6-hexachloro-2,4-cyclohexadien-1-one with solutions of [CoIII(apAr)2]–
forms CoIIICl(isqAr)2 (eq 1). The oxidation
state of the cobalt(III) center is unchanged in the 2e– bond-forming
reaction because both of the reducing equivalents derive from 1e–
oxidation of the o-amidophenolate
chela
The nucleophilic nature of the cobalt(III) center in [CoIII(apAr)2]–
is further revealed in reactions with alkyl halides. Addition of 1.0 equiv CH2Cl2
to [CoIII(apPh)2]– affords clean
conversion to the square pyramidal chloromethyl complex CoIII(CH2Cl)(isqPh)2
(Scheme 1). Reaction with CH3I gives a similar conversion to green
Co(CH3)(isqPh)2. Thus, the [CoIII(apAr)2]–
species have properties reminiscent of "supernucleophilic" cobaloxime(I)
complexes. The reaction with CH2Cl2 is a remarkable
example of nucleophilic attack on an unactivated alkyl halide under extremely
gentle conditions.
Addition of 1.0 equiv PhLi to CoIII(CH3)(isqPh)2
yields [CoIII(apPh)2]– and toluene.
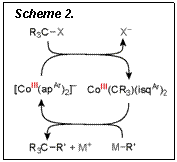
Such SN2-type pseudo-reductive elimination closes a catalytic cycle
for cross coupling with alkyl halides (Scheme 2).
Selective aerobic oxidations. We have reported the synthesis and characterization of the
manganese(III) anions [MnIII(X4cat)2(L)n]– [X = Cl, Br; n = 1, L = MeOH, OPPh3;
n = 2, L = acetone, tetrahydrofuran (THF)] as precursors to the square planar [MnIII(X4cat)2]–
core. The axial ligands are
substitution labile while the [MnIII(X4cat)2]–
core is preserved in non-aqueous
solutions. Previous reports had speculated that a vacant coordination site was
a prerequisite to reaction of [MnIII(X4cat)2(L)n]– complexes with O2.
However, we found that the [MnIII(X4cat)2]–
fragment reacts sluggishly with dioxygen.
The tris(catecholato) trianion [MnIII(X4cat)3]3–
is the true air-sensitive species, affording [MnIII(X4cat)2(L)2]– (L = acetone, THF) and X4bq with O2
exposure. The conversion of X4catH2
to X4bq is an aerobic oxidative dehydrogenation (2H+, 2e–)
reaction. We postulated that the [MnIII(Br4cat)2]–
core could catalyze
![Text Box: Figure 1. Solid-state structure of [CoIII(apiPr)2]– shown with 50% probability ellipsoids. Hydrogen atoms, CH3CN solvate and Cp*2Co+ countercation omitted for clarity.](images/Paper_9299_abstract_3812_0.gif)
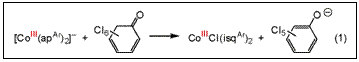
![Text Box: Scheme 1. Electrophilic Activation of CH2Cl2 by [CoIII(apAr)2]– to make CoIII(CH2Cl)(isqPh)2a aSolid state structure of Co(CH2Cl)(isqPh)2 shown with 50% probability ellipsoids. Hydrogen atoms on isqPh omitted for clarity.](images/Paper_9299_abstract_3814_0.gif)