46864-G7
Templating Ion-Conducting Membranes Using Block Copolymers
The need for clean and sustainable energy sources is growing rapidly,
and electrochemical devices such as batteries and fuel cells are considered
promising solutions. Polymer
electrolyte membranes for use in lithium batteries and fuel cells require high
ionic conductivities to decrease the internal potential losses. Besides ionic
conductivities, adequate mechanical strength is necessary to prevent
short-circuiting between electrodes and to reduce dendrite formation in lithium
batteries. Additionally, thermal
stability, chemical resistance, and controlled permeabilities To achieve the above requirements for lithium ion batteries, block
copolymers containing conducting blocks like poly(ethylene oxide), and
complementary blocks, have been employed to vary the morphology and properties of
conducting materials. The self-assembly of these copolymers permits the design
of flexible and sturdy membranes that contain conducting channels for ion
transport. Most
copolymer/salt doping has focused on the low salt concentration regime,
analogous to ether oxygen to lithium cation ([EO]:[Li]) ratios ranging from 50:1
to 12:1; however, recent literature has shown the promise in using higher salt
concentrations. For our
studies to date, we have employed cylinder-forming poly(styrene-b-ethylene oxide) [PS-PEO] block copolymers, where the polymer electrolyte cylinders were
formed via a self-assembly process inside a polystyrene matrix. We show that the domain spacing of our polymers increased as the degree
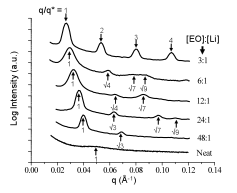
of salt-doping within the PEO domains increased. This trend is shown succinctly in the SAXS profiles in
Figure 1, where the primary scattering peak shifts to smaller q with increased salt. Additionally, the neat (non-doped) material,
along with the polymers at salt doping ratios ranging from 48:1 to 6:1, d*=2p/q*
(equation 1) The complete temperature dependent behavior of the PS-PEO:LiClO4
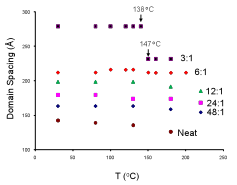
block copolymer complexes is shown in Figure 2. One particular point of interest is the discontinuity in
domain spacing for the 3:1 sample as the temperature is cooled from 147 oC
to 138 oC. This
discontinuity was linked to the crystallization of the 3:1 EO:LiClO4
complex in a recently published work. We note that the crystallization occurred
above the glass transition temperature of the PS matrix; however, the increased
segregation strength upon doping likely promoted the confined crystallization
of the PEO domains, leading to the larger domain spacing of the lamellae below
the discontinuity. Differences
in the domain spacing between copolymers doped lithium salts at similar ratios,
but with the various counterions, also were found in this study. The LiAsF6-doped polymer
exhibited the largest characteristic size, while the LiCF3SO3-doped
polymer showed the smallest domain spacing. These differences in domain spacing are likely related to
alkali metal – counterion interactions in the presence of the We
will continue to evaluate the influence of the lithium counterions on the
thermodynamics of block copolymer self-assembly, to understand and stabilize
conducting networks and explore the conductivity of these self-assembled
materials. Figure 1.In-situ
SAXS profiles of LiClO4-doped PS-b-PEO
as a function of salt concentration. Samples were held in the SAXS under vacuum
and at 1
Figure 2.(color online) Domain spacing vs temperature
calculated from in