Reports: ND455820-ND4: Theoretical and Experimental Investigations of Carbocation Diradicals
 printer friendly
printer friendlyThe textbook view of the electronic structure of a carbocation is that of an approximately sp2 hybridized carbon with an empty p orbital. This simplistic picture stands in sharp contrast to the related nitrenes, carbenes, nitrenium ions, oxenium ions and other reactive intermediates that possess one or more lone pairs of electrons that can be distributed between two orbitals, allowing (at least) two closed-shell singlet configurations, an open-shell singlet configuration, and a triplet configuration, each of which potentially offers up a unique landscape of reactivity and properties. Since carbocations do not possess a lone pair, and given early theoretical investigations by Schleyer suggested that simple carbocations had large energy gaps to open-shell states, the simple view of the electrophilic closed-shell singlet carbocation has largely remained unchallenged.
Here, we demonstrated that carbocations conjugated to the non-alternant hydrocarbon azulene can have low-energy or ground state triplets depending on the point of attachment. A detailed investigation into the origin of this spin state reversal, including NICS calculations, structural effects, substitution patterns, detailed linear free-energy relationships, and orbital analyses, allowed us to distill a set of principles that explains why these azulenyl carbocations have low-lying diradical states. These principles allowed for the prediction of a set of analogous structures containing not only carbocations, but also carbanions that exhibit low-lying or ground state triplets using simple canonical structure ideas. These examples point to not only a new and potentially broad class of carbocations that defy the textbook paradigm that carbocations are electrophilic species with closed-shell singlet ground states, but also to a new class of carbanions that may have application to high-spin materials.
The linear-free energy relationships for various substituted azulenyl-cations are shown below:
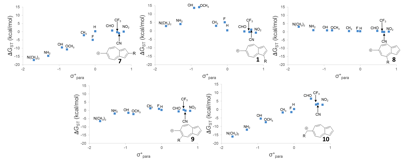 Linear free-energy relationships
of structures (UB3LYP/6-31+G(d,p)).
Linear free-energy relationships
of structures (UB3LYP/6-31+G(d,p)).
Understanding the origin of this remarkable spin inversion was performed by investigating the antiaromaticity using Nucleus Independent Chemical Shift calculations, shown in the figure below.
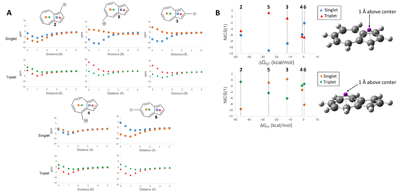
NICS values of the center of the 5-membered and 7-membered rings for structures 2-6 on the singlet and triplet surfaces. A) NICS value versus distance above plane of ring. B) NICS(1) value of 2-6 plotted against the ΔGST. (■ 5-Membered Ring Singlet, ▲ 5-Membered Ring Triplet, ● 7-Membered Ring Singlet, ♦ 7-Membered Ring Triplet) GIAO-HF/6-31+G(d,p)//CBSB7
From these calculations we were able to predict a broad class of ions having low-energy diradical states. Some of these are shown in the figure below:
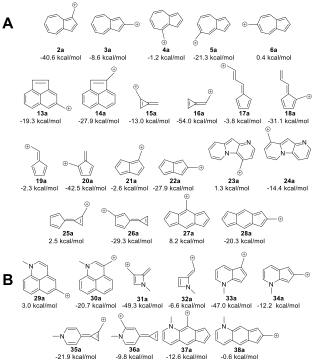
ΔGST of additional ions. Ions with formally Huckel antiaromatic/Baird aromatic canonical structures have low-lying diradical states. B) Manipulation of the ΔGST through the introduction of a 3° amine leads to a change in the favored spin state. UB3LYP/6-31+G(d,p)
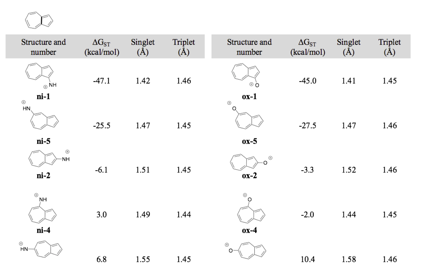
This work was then shown to be a general phenomenon, as seen
with azulenyl nitrenium and
oxenium ions:
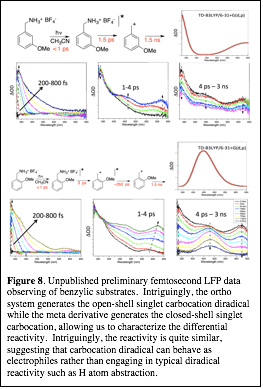
Experimentally, we have tried to detect the triplet carbocation from formation of the m-methoxy carbocation—this carbocation is predicted to have nearly degenerate singlet and triplet states. However, the mechanism currently appears to provide the singlet carbocation, which undergoes normal carbocation decay mechanisms. This supports the singlet ground state of this structure. Currently, we are working on directly detecting a triplet carbocation and characterizing its decay pathways.
| Figure 8. Unpublished preliminary femtosecond LFP data observing of benzylic substrates. Intriguingly, the ortho system generates the open-shell singlet carbocation diradical while the meta derivative generates the closed-shell singlet carbocation, allowing us to characterize the differential reactivity. Intriguingly, the reactivity is quite similar, suggesting that carbocation diradical can behave as electrophiles rather than engaging in typical diradical reactivity such as H atom abstraction.
|