Reports: ND456665-ND4: Paracyclophane Self-Assembly Promoted by Transannular Hydrogen Bonding
 printer friendly
printer friendlyBackground and Significance
[2.2]Paracyclophane ([2.2]pCp) is a molecule of historic intrigue due to its unique structural, chemical, physical, and optical properties – these include non-planar aromaticity, through-space conjugation, planar chirality, and rigidity. In fact, the unambiguous and stereochemically defined structure has led to it being referred to by H. Hopf as a “hexadecavalent ‘superatom’” onto which 16 substituents may be introduced, and “a core element to organize space”.1 While [2.2]pCp has been used to great effect in ligand design and conjugated polymers, it has been relatively underexplored in supramolecular constructs. With this realization, we recently reported a new derivative, [2.2]pCp-4,7,12,15-tetracarboxamide, [2.2]pCpTA ((±)-1, Figure 1), which advantageously utilizes hydrogen bonding (H-bonding) to form supramolecular 1-D “nanorods”.2 The design exploits the first pCp transannular H-bonding (to preposition the amide groups for intermolecular hydrogen bonding) and planar chirality of the monomer (to dictate the chirality of the 1-D assembly). Also noteworthy is that the dipole moment of the monomers can cancel upon nanorod growth (which we presume affects the assembly mechanism), and the R group can be conveniently varied. The overlapped, multi-tiered aromatic core of the suprastructure is attractive for materials science applications involving charge transport. Described here is our progress associated with the two specific aims of the funded proposal: (1) The design, synthesis, and resolution of pCps capable of transannular H-bonding for the evaluation of the consequences of these interactions on molecular pCp structure, stability, and chiroptical properties and (2) the study of the transannular H-bonding promoted solution-phase self-assembly and emergent electronic structures of selected pCps.
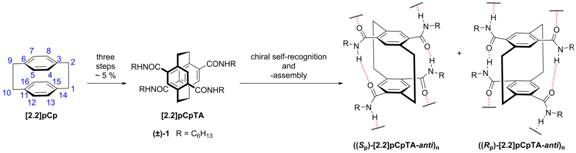
Figure 1. The parent [2.2]pCp is functionalized with four amides to form [2.2]pCpTA, which self-assembles via cooperative transannular and intermolecular hydrogen bonding.
Investigation of Transannular Hydrogen Bonding
In order to understand the strength and influence of transannular hydrogen bonding on the self-assembly of pCp carboxamides, model compounds 4,16-bis(amide)[2.2]pCp 2 (pseudo-para (ps-p)) and (±)-4,12-bis(amide)[2.2]pCp (±)-3 (pseudo-ortho (ps-o)) were synthesized from the corresponding acids during this reporting period.3 Compounds 2 and (±)-3 serve as the non-intramolecular hydrogen-bond control and transannular hydrogen-bonding model, respectively. These derivatives show remarkably different amide N–H 1H NMR resonances in CDCl3 (Figure 2), consistent with their expected hydrogen bond capabilities; the relative N–H chemical shift of 2 (5.5 ppm) is shifted significantly downfield for (±)-3 (7.4 ppm). This deshielding effect is due entirely to the presence of the transannular hydrogen bond in (±)-3, which is absent in 2. With the synthetic technology in hand to prepare these bisamides, we are now prepared to evaluate their self-assembly properties and use them to establish chiral resolution conditions.
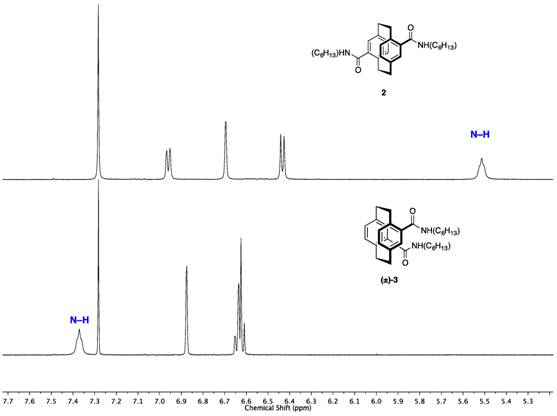
Figure 2. 1H NMR of pseudo-para [2.2]pCp bisamide 2 (top) and pseudo-ortho [2.2]pCp bisamide (±)-3 (bottom) at 30 mM in CDCl3.
Chiral Resolution
Enantiopure monomers are needed in order to fully study the pCp self-recognition process, and thus, the chiral resolution of (±)-1 and transannular H-bond comparator (±)-3 is underway. Attempts to resolve (±)-4 (Figure 3a) via derivatization with a chiral alcohol to form separable diastereomers proved troublesome; esterification does not proceed as expected under Fischer conditions or in the presence of a dehydrating agent.
Resolution of a pseudo-ortho comparator has been accomplished by adapting a method developed by Morisaki, et. al. (Figure 3b).4 Accordingly, (±)-6 could be converted into separable sulfinate diastereomers (Rp,S)-7 and (Sp,S)-7. Subsequent lithiation and CO2 quench offered optically pure diacid (Rp)-8. Following our already established synthetic methodology from here, we should be able to synthesize enantiopure pseudo-ortho bisamide (Rp)-3. In parallel, we are currently using this procedure to access enantiopure [2.2]pCpTA (Figure 3c).
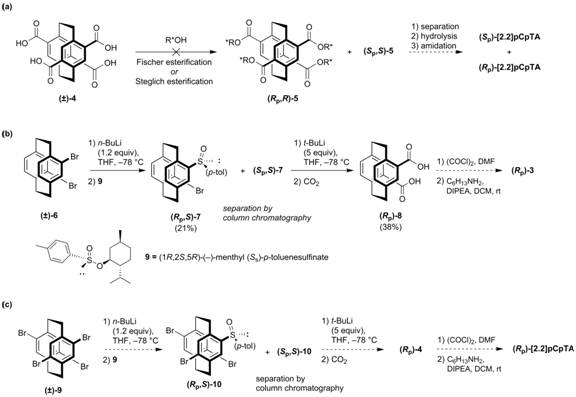
Figure 3. (a) Attempted chiral resolution of (±)-[2.2]pCpTA through derivatization, (b) resolution of (±)-3 through conversion to chiral sulfinates, and (c) planned resolution procedure for (±)-[2.2]pCpTA.
[3.3]Paracyclophane: A Strain-Relieved Scaffold
The strain of [2.2]pCp arises from ring distortions and repulsive arene-arene interactions. We originally hypothesized that the reduced strain enthalpy of [3.3]paracyclophane, [3.3]pCp (Figures 4a and 4b), signified by a more reasonable interplanar deck distance and flatter benzene rings, should favor formation of the H-bonded assembly. Unlike [2.2]pCp, [3.3]pCp is not commercially available and must be synthesized before introducing the methodology developed for [2.2]pCpTA.5 While the derivatization of [3.3]pCp differs from that of [2.2]pCp, with significant optimization (Figure 3c), (±)-[3.3]pCpTA could be obtained. 1H NMR analysis shows the amide N–H resonance of (±)-[3.3]pCpTA occurs downfield at 7.4 ppm in CDCl3, similar to that of (±)-[2.2]pCpTA and (±)-3. This observation indicates that the transannular hydrogen bond is indeed formed in (±)-[3.3]pCpTA, and the desired assembly will likely be accommodated. We are currently scaling up the synthesis and purification of this compound to allow its characterization in the solid-state and solution.
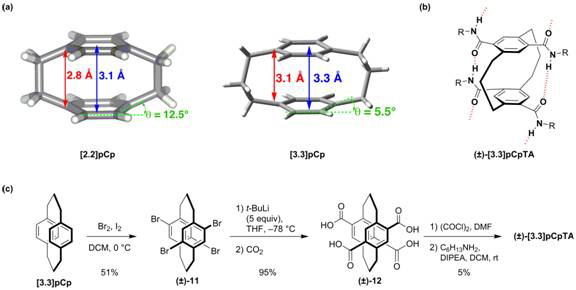
Figure 4. (a) X-ray crystal structures of [2.2]- and [3.3]-paracyclophane with transannular deck distances and benzene deformations shown, (b) chemical structure of (±)-[3.3]pCpTA, and (c) synthesis of (±)-[3.3]pCpTA.
References
1. Henning Hopf, Isr. J. Chem. 2012, 52, 18–9
2. Fagnani, D. E.; Meese, M. J., Jr.; Abboud, K. A.; Castellano, R. K. Angew. Chem. Int. Ed. 2016, 55, 10726–10731
3. D. Y. Antonov, E. V. Sergeeva, V. I. Rozenberg, Russ. Chem. Bull., 1997, 46 1897–1900
4. Y. Morisaki, Y. Chujo, Chem. Lett. 2012, 41, 990–992
5. M. Shibahara, M. Watanabe, T. Iwanaga, T. Matsumoto, K. Ideta, T. Shinmyozu, J. Org. Chem., 2008, 73, 4433–4442










