Reports: DNI756281-DNI7: Designing the Liquid-to-Solid Transition in Polyelectrolyte Complexes
 printer friendly
printer friendlyWe proposed the design of self-assembling polymer-based materials engineered to exhibit a wide range of mechanical properties. We focus on the modulation of orthogonal interactions in a class of materials based on polyelectrolyte complexation to achieve this goal. Polyelectrolyte complexes (PECs) are a unique class of polymeric materials that can manifest in both liquid and solid forms (Figure 1). Complex coacervation refers to the associative liquid-liquid phase separation resulting from the complexation of oppositely-charged polyelectrolytes, and is thought to occur as a result of an initial electrostatic attraction, followed by an entropically-favored release of counter-ions and water. A similar process can also lead to the formation of solid precipitates, which are analogous to polyelectrolyte multilayer films formed using layer-by-layer deposition. While the most significant differences that determine the formation of a solid vs. a liquid complex are thought to be the strength of intermolecular interactions and the extent to which salt and water are excluded from the final complex, an understanding of the fundamental material transitions between solid and liquid phases is critically absent.
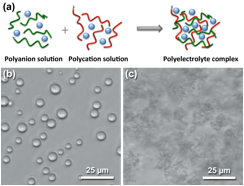
Figure 1. (a) Schematic depiction of polyelectrolyte complex formation. Optical micrographs of (b) complex coacervates and (c) solid precipitates formed from a mixture of oppositely-charged polymers.
Our work utilizes rheological analysis to clearly identify the nature of the liquid-to-solid transition. Spectroscopic rheological analysis of liquid samples near the phase transition can be used to clearly distinguish gelation from a glass transition. To date, we have explored the effect of electrostatic interactions on this liquid-to-solid transition in PECs by varying the concentration and identity of salt present. We are currently engaged in understanding the effect of polymer chain length and hydrophobicity on the PEC phase behavior and the resulting material properties.
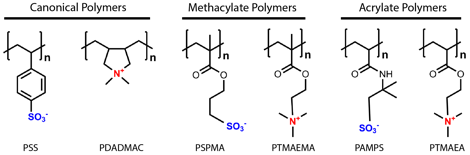
Figure 2. Chemical structure of the canonical polyelectrolyte complex system poly(4-styrenesulfonic acid) (PSS) and poly(diallyldimethyl ammonium) (PDADMAC), as well as the model methacrylate-based polyelectrolytes poly(3-sulfopropyl methacrylate) (PSPMA) and poly(2-(N,N,N-trimethyl amino)-ethyl methacrylate) (PTMAEMA), and the analogous acrylate polyelectrolytes poly(2-acrylamido-2methylpropane sulfonic acid) (PAMPS) and poly(2-(N,N,N-trimethyl amino)-ethyl acrylate (PTMAEA).
Our initial experiments focused on the salt-driven liquid-to-solid transition in the canonical PEC system of poly(4-styrenesulfonic acid) (PSS) and poly(diallyldimethyl ammonium) (PDADMAC, Figure 2). We combined direct measurement of PEC phase behavior with a time-salt superposition analysis of the linear viscoelasticity of the materials (Figure 3). This superposition allows for extension of the range of timescales over which the viscoelastic response is measured from two decades in frequency space to nearly ten. Furthermore, direct comparison of the measured salt and polymer concentration in the coacervate with the subsequent rheological character enables a more generalizable description of these materials.
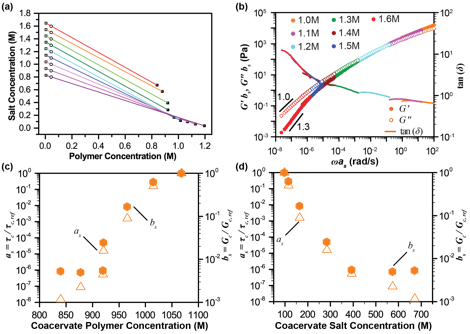
Figure 3. (a) Coacervate binodal curve as a function of polymer concentration and salt concentration. Open circles indicate the ‘as prepared’ concentration of the overall samples. Tie lines connect these initial conditions with the experimentally measured concentrations for the supernatant (open squares) and coacervate (solid squares). Polymer concentration is expressed on a monomer basis. (b) Plot of the time-salt superposition of frequency sweep data for complex coacervates, shifted with respect to the 1.0M ‘as prepared’ data. Plots of the horizontal (open triangles) and vertical (solid hexagons) shift factors, as a function of the (c) measured polymer and (c) salt concentration in the coacervate phase.
Further analysis of the viscoelasticity of these materials demonstrated that decreases in the salt and water content of PSS/PDADMAC complexes results in the formation of a physical gel, where the network forms as a result of trapped electrostatic crosslinks that percolate the sample at a critical salt concentration. This gel point was unambiguously identified based on the condition where a frequency-invariant response was observed (Figure 4a). Additional analysis of the data (Figures 4b,c) show excellent agreement with theoretically expected trends.
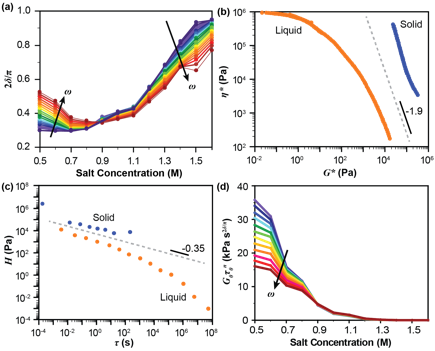
Figure 4. (a) A plot of normalized phase angle (2δ/¹) vs. salt concentration for varying frequencies (purple to red), indicated by the arrows. The gel point occurs at 0.85M KBr and 2δ/¹ = 0.35, based on the frequency-invariant crossover point. (b) A plot of complex viscosity (η*) vs. complex modulus (G*) for liquid and solid complexes resulting from the time-salt superposition data. The dashed grey line with slope of 1.9 indicates the predicted divergence line with the expected power-law behavior at the gel point. (c) A plot of the relaxation time spectrum (H) vs. relaxation time (t) showing the characteristic decrease in the relaxation time spectrum at long times for liquids (orange) and suggesting a gel-like plateau in the relaxation time spectrum for solid samples (blue). The dashed grey line with slope of 0.35 indicates the expected power-law relaxation behavior at the gel point. (d) A plot of the front factor G0t0n vs. salt concentration for varying frequencies (purple to red).
Looking forward, we will expand our studies to characterize the role of polymer chemistry and chain length. This work will continue to utilize PSS/PDADMAC, but will also expand to include two additional polymer systems – one based on a methacrylate backbone, and the other on an acrylate structure (Figure 2). We have already synthesized the methacrylate polymers with degree of polymerization N = 50, 250, and 450 using controlled polymerization, in collaboration with Prof. Todd Emrick's lab at the University of Massachusetts Amherst. Low polydispersity acrylate polymers with N = 50, 250, 1000, and 2000 were provided by Prof. Brent Summerlin's lab at the University of Florida. These three systems will allow for a direct comparison of the effect of small changes in backbone chemistry, as well as larger changes in the hydrophobicity of the polymers. Furthermore, the methacrylate chemistry will allow for the straightforward incorporation of additional chemical functionality for future experiments geared at investigating the effects of hydrogen bonding and/or zwitterion-driven hydration as copolymers to enable the independent control of material properties in new classes of polyelectrolyte materials. By relating material properties to polymer structure and bonding effects, we will have the potential to design material properties by varying polymer chemistry and architecture. This knowledge will allow for the use of polyelectrolyte complexation as a new class of materials across a wide range of applications.










