Reports: UR156502-UR1: Molecular Jump Rope: Synthesis and Study of Axial Chirality in Cyclophanes Lacking Stereocenters
 printer friendly
printer friendlyIntroduction
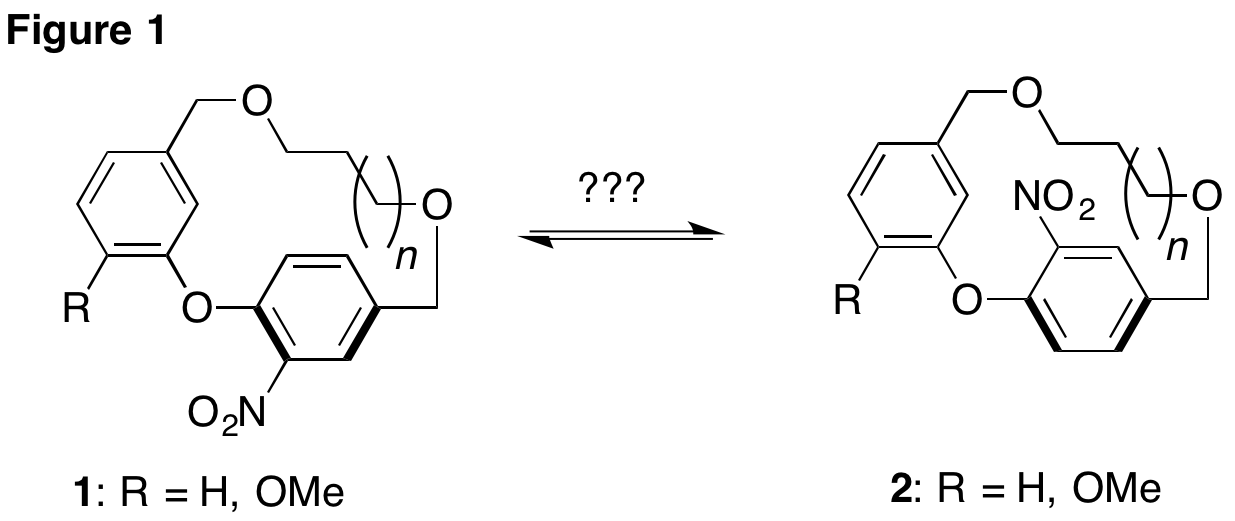
In our research plan, we proposed the synthesis of a series of diaryl ether paracyclophanes with varying tether lengths (Figure 1, n=0-5). We also stated that at the end of year one, the synthesis of these nitro substituted cyclophanes would be completed, and qualitative 1H-NMR data will allow for the identification of tether lengths that render these cyclophanes chiral at room temperature. As is often the case in the research lab, unforeseen obstacles have prevented the preparation of these cyclophanes, but what we have learned from these setbacks will no doubt be of use as we move forward toward our goal. After one year, it is clear that this is a challenging problem, but one that we are confident that we are equipped to solve. The results presented here will show that we are on the brink of developing a scalable, rapid, and modular synthesis of various cyclophanes that will be of use to study the chirality of these molecules. As stated in the proposal, these unique cyclophanes will be the starting point for the development of new chiral bisphosphine ligands in future years of the grant.
 Synthetic Efforts in Year 1
Synthetic Efforts in Year 1
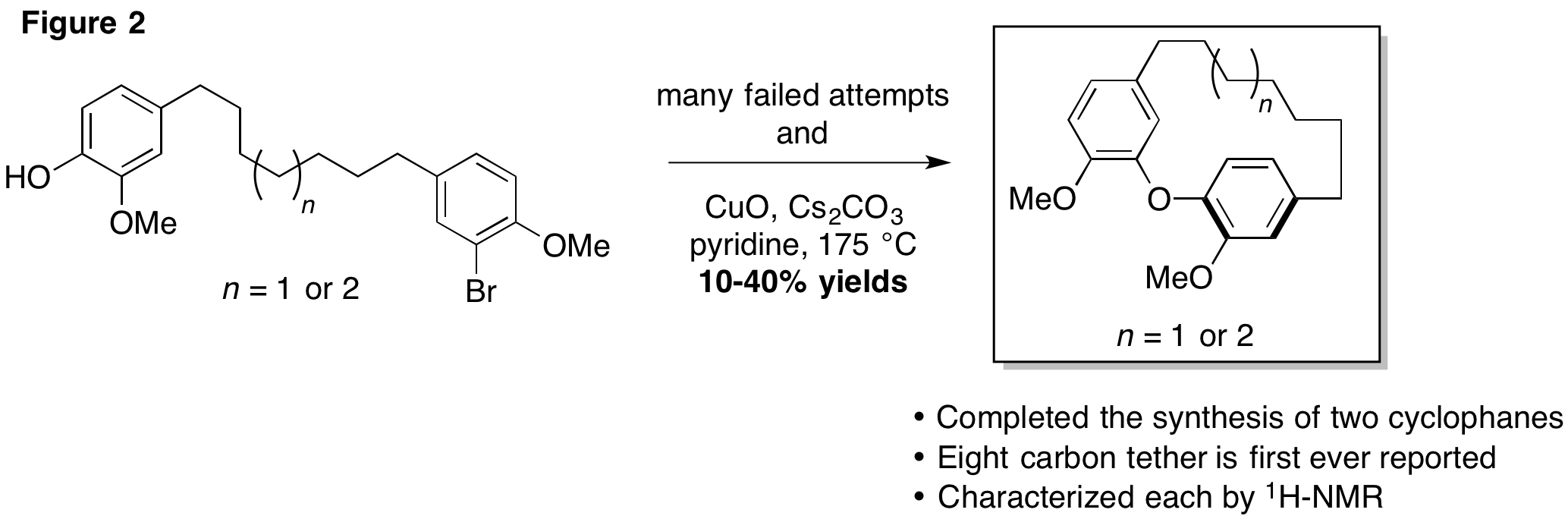
As mentioned in our original proposal, we began by pursuing an Ullmann coupling as the key cyclophane forming step, as illustrated in Figure 2. We identified several deficiencies with this approach which necessitated a change of strategy. The cyclization was low yielding and each substrate required at least six steps to prepare.
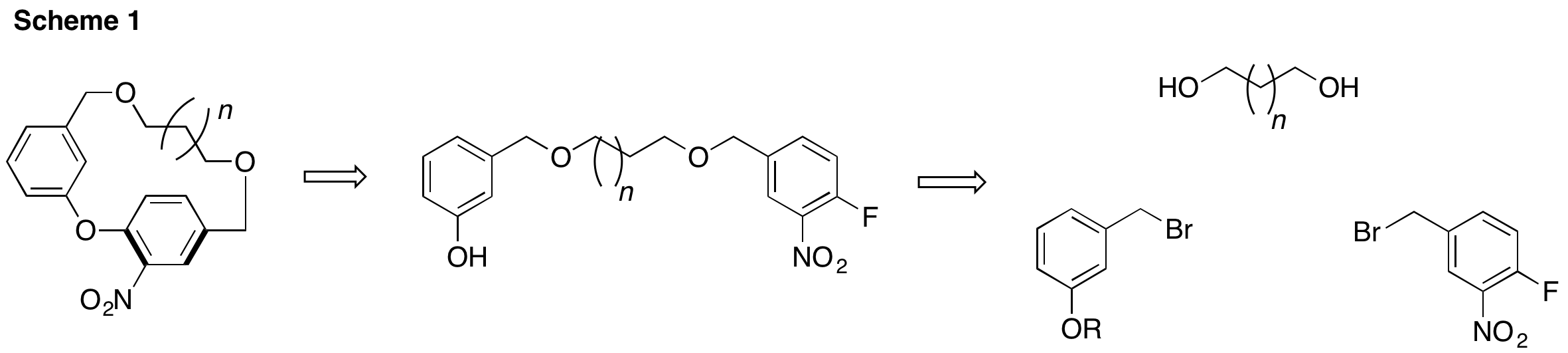 In light of these results, this year we pursued a new approach that attempted to address these deficiencies. We envisioned that two alkyl halide building blocks could be mass produced and could be appended to varying diols through straightforward, successive Williamson ether syntheses. Finally, cyclophane closure would be accomplished via a key SNAr cyclization based on a high yielding analogous reaction reported in the literature. A key to pursuing this strategy would be to identify an appropriate protecting group for the phenol moiety that would survive the Williamson ether synthesis but could be removed prior to the key SNAr cyclization.
In light of these results, this year we pursued a new approach that attempted to address these deficiencies. We envisioned that two alkyl halide building blocks could be mass produced and could be appended to varying diols through straightforward, successive Williamson ether syntheses. Finally, cyclophane closure would be accomplished via a key SNAr cyclization based on a high yielding analogous reaction reported in the literature. A key to pursuing this strategy would be to identify an appropriate protecting group for the phenol moiety that would survive the Williamson ether synthesis but could be removed prior to the key SNAr cyclization.
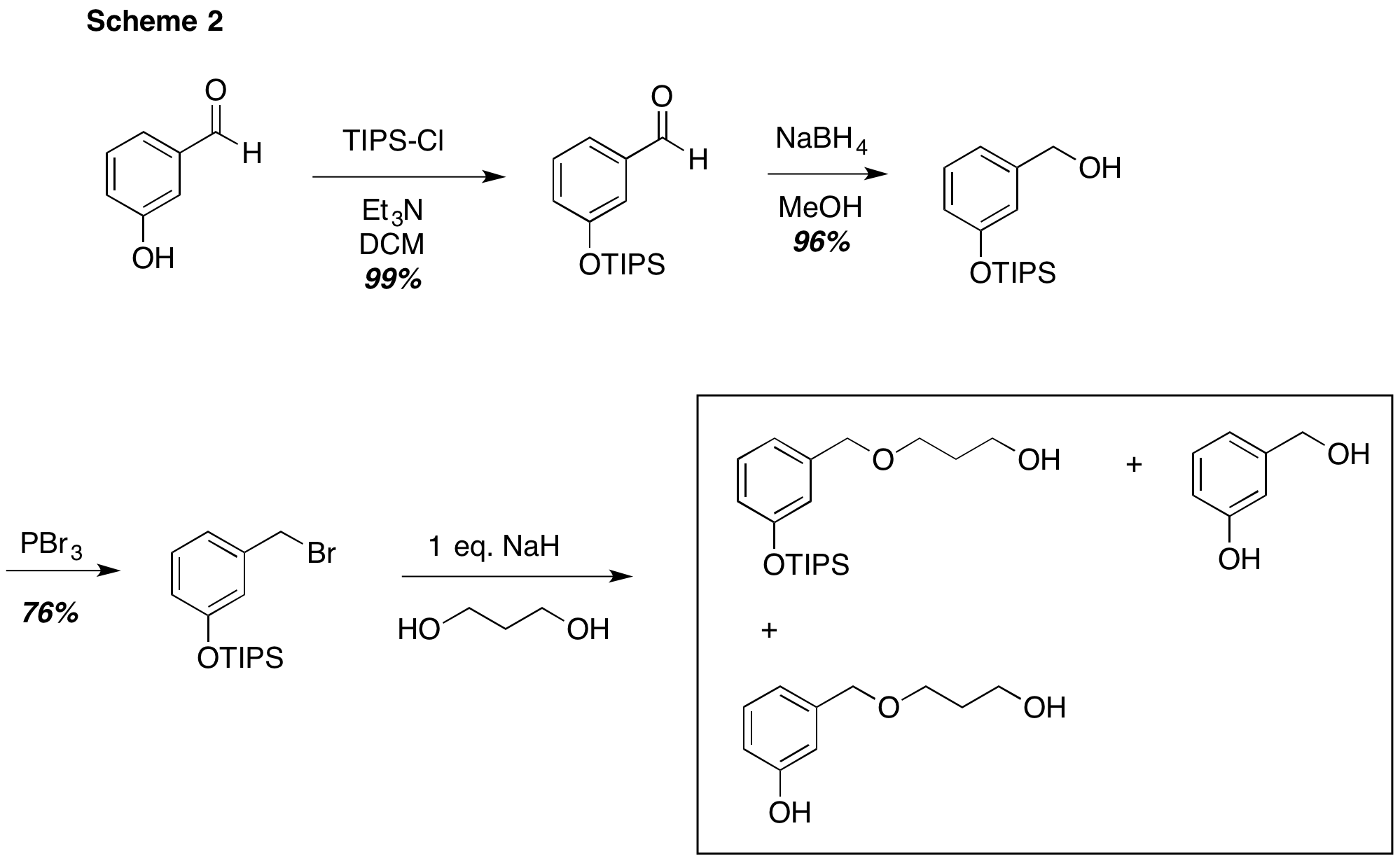 We first tried using a bulky TIPS protecting group to shield the phenolic oxygen from competitive alkylation. As shown in Scheme 2, introduction of the silyl ether, reduction of the aldehyde and transformation to the corresponding bromide were uneventful. However, the strongly basic conditions required for the Williamson ether synthesis led to a complex product mixture that consisted of the desired product as well as various phenolic compounds. Other approaches that are not pictured in Scheme 2 included switching the protecting group to a more stable SEM group and attempting to employ a trichloroacetimidate leaving group under acidic conditions. Neither of these approaches was productive.
We first tried using a bulky TIPS protecting group to shield the phenolic oxygen from competitive alkylation. As shown in Scheme 2, introduction of the silyl ether, reduction of the aldehyde and transformation to the corresponding bromide were uneventful. However, the strongly basic conditions required for the Williamson ether synthesis led to a complex product mixture that consisted of the desired product as well as various phenolic compounds. Other approaches that are not pictured in Scheme 2 included switching the protecting group to a more stable SEM group and attempting to employ a trichloroacetimidate leaving group under acidic conditions. Neither of these approaches was productive.
Next, we attempted to utilize the much more stable methyl ether protecting group.
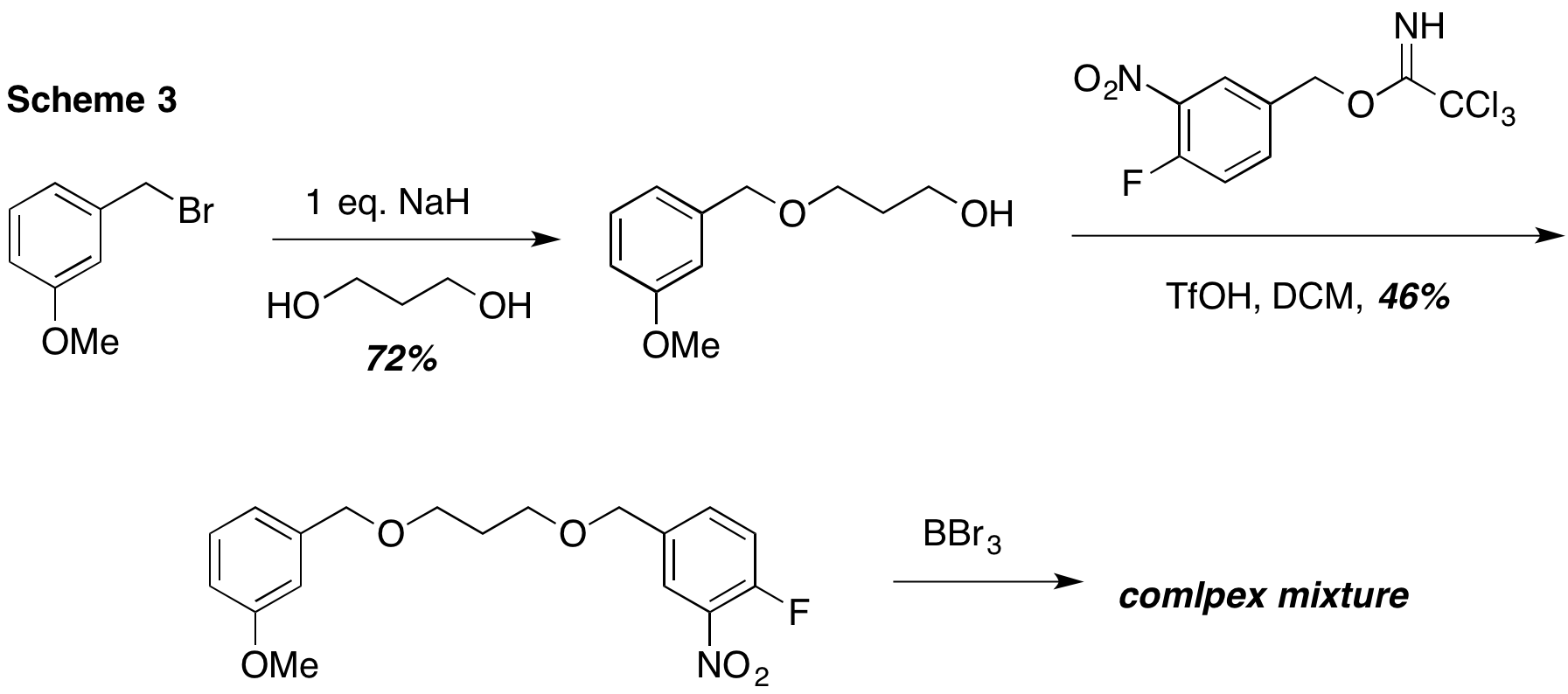
Beginning with commercially available 3-methoxy benzyl bromide, alkylation led to the mono alkylated 1,3-propandiol shown. A second alkylation was carried out using the trichloroacetimidate to avoid reaction of the fluorinated aryl ring under basic conditions. This led to a modest yield of the differentially dialkylated 1,3-propandiol. However, while several literature examples suggested that methyl and benzyl ethers could be differentially deprotected using BBr3, that was not our experience. One or more benzyl ether bonds were severed using this string Lewis acid. Once again, it appeared that we needed to reexamine our approach.
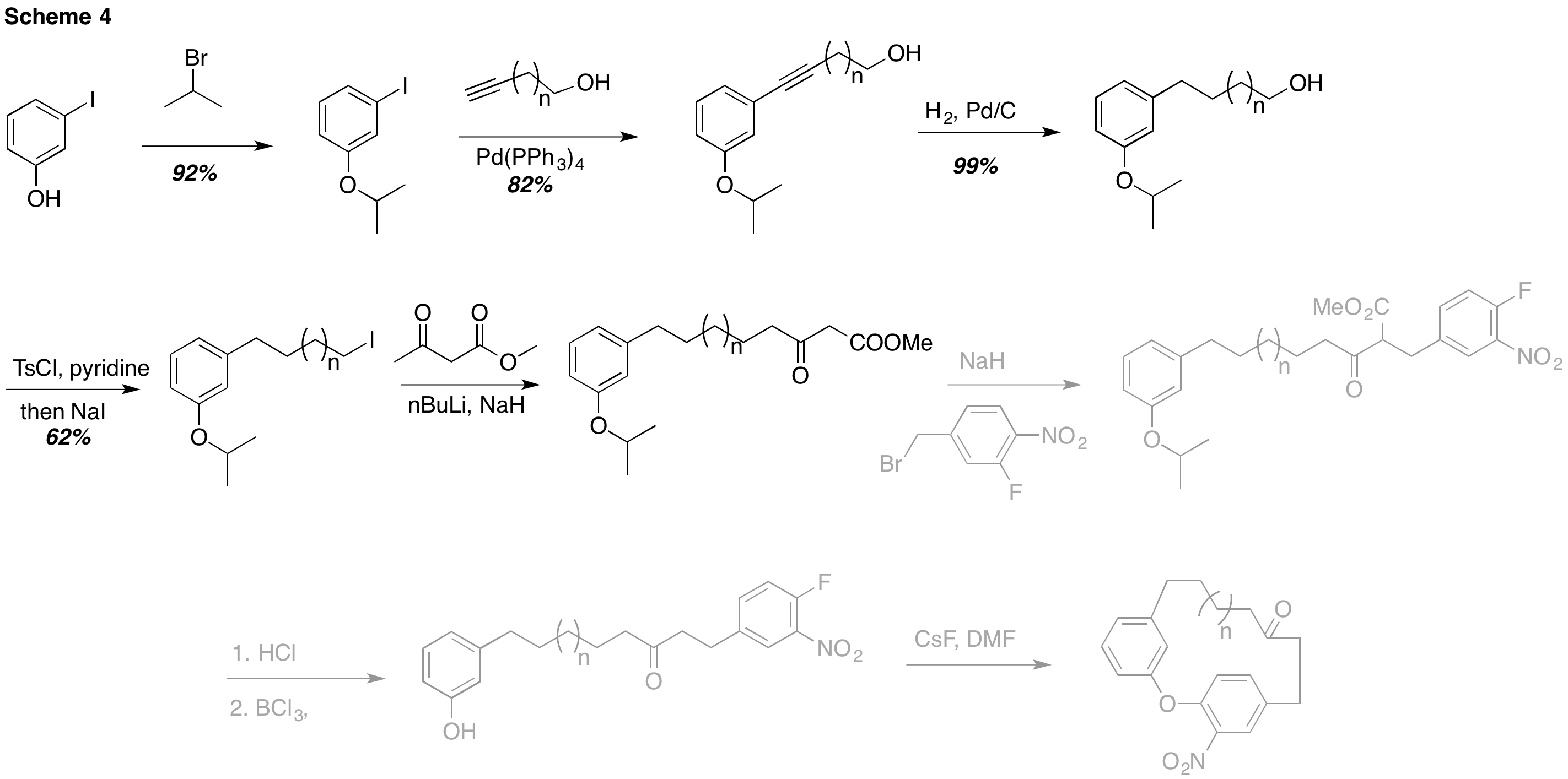
Hoping to gain confidence, we adapted an established literature procedure to produce the SNAr substrates. Our progress is shown in Scheme 4. Following previous approaches, we utilized an iso-propyl ether as a protecting group. Sonogashira coupling and hydrogenation allowed us to make various primary alcohols of varying lengths. Next, tosylation and iodination led to the corresponging iodide. However, displacing the iodide with the dianion of methylacetoacetate was not successful after multiple attempts that included varying time, temperature, and concentration.
Plans in the Coming Year
After a year of funded work, we have learned a great deal about which approaches are the most advantageous. Based on the difficulty associated with adding and removing protecting groups, we will seek out a protecting group free approach to these cyclophanes in the coming year. In addition, sequences requiring 5 or more steps are too time and labor intensive for undergraduate students, even if they are devoting their full time and attention to these problems in the summer months. On the positive side, we have had great success with the Sonogashira reaction to append the aromatic ring to the variable tether. We would also like to continue pursuing an SNAr ring closing as the key cyclophane forming step. Thus, our ideal approach going forward would include these criteria:
· 3-4 steps to make each cyclophane from commercially available materials
· Utilize the Sonogashira reaction to append aromatic rings
· Protecting group free
· SNAr ring closing to form all cyclophanes
· Allow for flexibility in the tether length
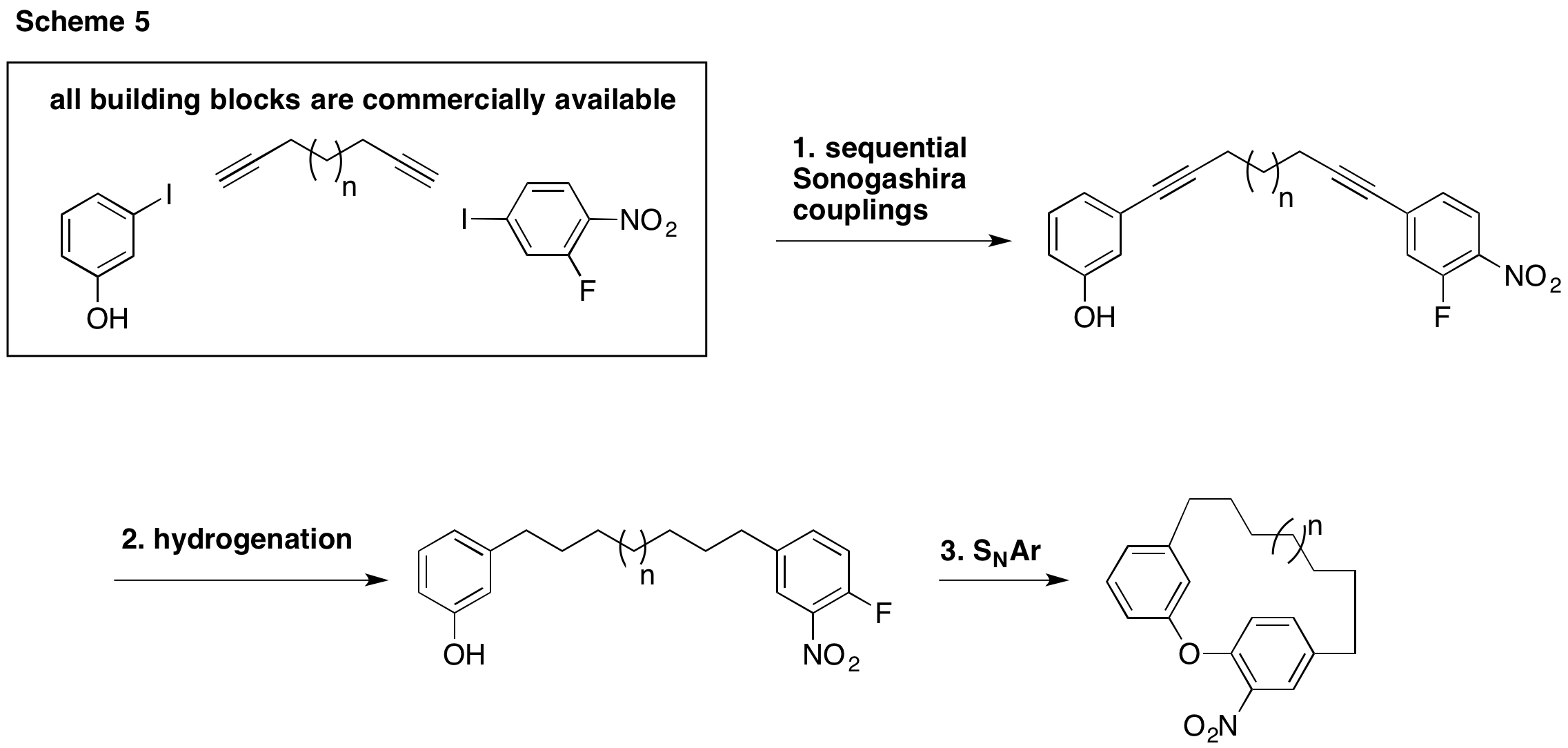 With these criteria in mind, we have begun a new and improved approach to making the cyclophanes we wish to study. We plan to couple 3-iodophenol with various terminal dialkynes (1,6-heptadiyne, 1,7-octadiyne, etc) followed by coupling with 4-iodo-2-fluoronitrobenzene. Hydrogenation and cyclization would complete each cyclophane. Should competitive hydrogenation of the nitro group be an issue, a simple mCPBA oxidation would restore this key functionality. Since the first three steps are palladium catalyzed, one could also imagine the development of a one-pot procedure.
With these criteria in mind, we have begun a new and improved approach to making the cyclophanes we wish to study. We plan to couple 3-iodophenol with various terminal dialkynes (1,6-heptadiyne, 1,7-octadiyne, etc) followed by coupling with 4-iodo-2-fluoronitrobenzene. Hydrogenation and cyclization would complete each cyclophane. Should competitive hydrogenation of the nitro group be an issue, a simple mCPBA oxidation would restore this key functionality. Since the first three steps are palladium catalyzed, one could also imagine the development of a one-pot procedure.
Conclusion
The previous year has led us to revise our approach to the synthesis of varying cyclophanes. This new approach is protecting group free and streamlined. We expect to complete the synthesis of several cyclophanes in the coming year.










