Reports: UR751997-UR7: Photochemical Control of Nanoparticle Aggregation
 printer friendly
printer friendlyIn this report, we report results on the conformational dynamics of cis-azobenzene that has fluorine substitution at the ortho position of the aromatic rings. The use of azozbenzene (AB) isomerization as a molecular probe of cavity dimensions and polarity is an impetus for this work. AB is arguably the most studied organic chromophore in chemistry. AB exists as two geometric isomers with the trans conformation 12 kcal/mol more stable than the cis isomer. We have observed through-space (TS) 19F-19F coupling in the cis-conformation of ortho-fluoro azobenzenes. We argue that this coupling provides a sensitive probe of conformation of cis-AB in hybrid systems through the use of 19F NMR.
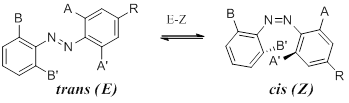
Scheme 1. Cis-trans (E-Z) isomerization of ortho-substituted azobenzene; library of compounds.
1 (F4OMe): A,A’,B,B’ = F; R=OMe
2 (Fa1Fb2OMe): A,A’,B’ = F; B = H; R=OMe
3 (Fb2OMe): A,A’ = F; B,B’ = H; R=OMe
4 (Fa1OMe): A,A’,B’ = H; B = F; R=OMe
5 (Fa2Fb1OMe): A,B,B’ = F; A’ = H; R=OMe
6 (Fb1OMe): A,A’ = F; B,B’ = H; R=OMe
7 (Fa1Fb1OMe): A,A’,B’ = H; B = F; R=OMe
8 (Fa2Fb1): A,B,B’ = F; A’ = H; R=H
Azobenzene compounds 1–8 (Scheme 1) were prepared via the diazonium salt for 1-7; 8 was prepared from 2-fluoronitroso benzene and 2,6-difluoroaniline. As prepared, the configuration of these compounds is principally Z, which can be converted to a mixture of Z and E in min using UV irradiation. The thermal Z to E isomerization is sufficiently slow so that isomer separation is possible by flash chromatography (the more polar Z elutes after E).
The 19F{1H} spectrum of (E)-1-(2,6-difluoro-4-methoxyphenyl)-2-(2,6-difluorophenyl)diazene is shown in Figure 1. Z1 exhibits two triplets with Jobs = 5 Hz. Table 1 lists the 19F chemical shifts for 1-8. All 19F spectra were collected with broadband proton decoupling. The fluorines on the methoxy-substituted ring are designated Fa. With the exception of 1 (Fa shift upfield by 0.2 ppm), the fluorines are shifted downfield upon photoisomerization to cis. No through-space coupling was observed for the trans isomer. We confirmed that the observed coupling is due to 19F-19F interaction via COSY and 2-D J-exchange experiments.
Variable temperature NMR was conducted to investigate dynamic behavior. No significant change was observed in toluene-d8 up to 98°C. At low temperatures, 1-3 and 7 displayed a decrease in the coupling with the multiplet coalescing near -100°C. We have assigned phenyl rotation as the dynamic process responsible for this diminution of through-space coupling.
Table 1. 19F NMR chemical shifts for 1-4, CDCl3, 28°Ca
Compound |
δ, ppm trans |
19F NMR δ, ppm cis |
|||
A,A’ (Fa) |
B,B’(Fb) |
A,A’ (Fa) |
B,B’(Fb) |
JFF, Hz |
|
1 |
-120.3 |
-125.2 |
-120.1 |
-122.6 |
4.82 |
2 |
-123.4 |
-125.0 |
-121.8 |
-123.0 |
4.08 |
3 |
|
-125.2 |
|
-123.0 |
|
4 |
-124.4 |
|
-120.9 |
|
|
5 |
-120.5 |
-127.5 |
-120.3 |
-125.46 |
4.53 |
6 |
|
-127.3 |
|
-125.4 |
|
7 |
-124.0 |
-128.0 |
-120.8 |
-125.0 |
2.22 |
8b |
-126.8 |
-124.4 |
-125.6 |
-122.4 |
5.90 |
a δreferenced to internal standard, C6F6 (-164.9 ppm). b For 8, Fa = methoxy substituted ring.
X-ray Analysis
We were able to obtain X-ray structures for Z1 and Z2 (Figure 2). The structural features of the optimized structure (b3ylp/cc-pVTZ) compared closely to the X-ray structure with bond lengths and angles all within 2%.
Density Functional Theory
The conformational behavior of Z1, Z2, Z5, and Z7 was investigated by density functional theory (DFT) using Gaussian09. It is has been noted that DFT on conjugated systems requires electron correlation. The most consistent and sensible results were obtained using the b3ylp method and the cc-pVTZ basis set. Calculations were performed without solvent fields; for 1, the four possible conformers were energetically equivalent and two transition states were found with a bisected geometry (see representation for 8 in TOC). For 1, the energy of the bisected transition state is 4.7 kcal above the ground state which compares favorably with the experimental value of 3 kcal (taken from the coalescence temperature in the NMR spectrum). This is the first experimental measurement of phenyl rotation in cis-azobenzene.
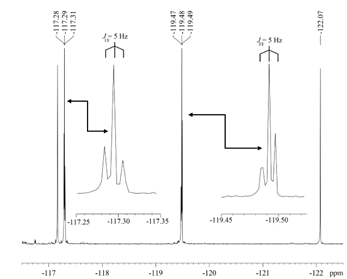
Figure 1. 19F{1H} NMR spectrum of E/Z in CDCl3 at 28°C for azobenzene 1.
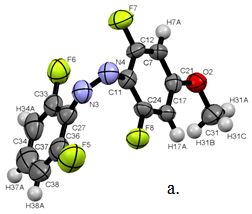
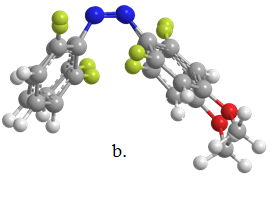
Figure 2. a. X-ray structure of Z1, b. overlap of X-ray and DFT structures for Z1.