Reports: DNI352704-DNI3: Designing Multifunctional Complexes for Chemical Catalysis
 printer friendly
printer friendlyPrior to the reporting period, we communicated the synthesis of a tris(diketimine) cyclophane ligand (H3L) and a hexacarboxamide cryptand (H6L’), which both template trinuclear clusters. This designed approach to clusters using macrobicycles was demonstrated in the synthesis of an (FeCl2)3 cryptate complex, as well as that of the triiron(II) tri(bromide) and a trimanganese(II) tri(bromide) cyclophane complexes. We have primarily focused on complexes of the cyclophane ligand. One of our goals for the proposal is to understand the role of metal ion type on reactivity. Therefore, we synthesized the corresponding trichromium(II), tricobalt(II), trinickel(II), tricopper(I), trizinc(II), trigermanium(II), tritin(II), and trialuminum(III) complexes using the cyclophane ligand (Figure 1).
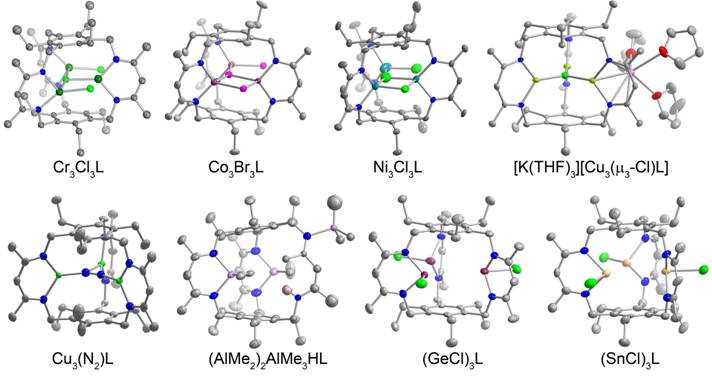
Figure 1.
Our results on copper cluster chemistry are summarized here. We reported the anionic chloride-bridged tricopper(I) compound, which exhibits strong luminescence upon excitation in the near UV. By using Cu(OTf) as our metal source, we were able to crystallize the first molecular complex containing Cu–N2 interactions, i.e., Cu3(N2)L. This adduct is possible because of the unparalleled size and shape selectivity afforded by our cyclophane; the steric constraints exclude solvent ligands to the metal centers, leaving N2 as the only available ancillary donor. From vibrational spectroscopy, the N–N stretching mode is lower in energy than expected for an unactivated N2 (i.e., 1952 cm-1 for 14N2). Computationally, minor changes to the orientation of the ligand arms results in a surprisingly large decrease (~200 cm-1) for the N–N vibration. This result is important because it shows that N2 reduction using Cu may be possible despite the poor π-backbonding for Cu. The fluorescence of this N2 adduct differs significantly from the chloride-bridged complex, and the guest dependent luminescence is currently under investigation. The N2 complex, Cu3(N2)L, readily reacts with S8 to afford the first example of a coordinatively unsaturated sulfide-bridged copper cluster in which all metal centers are in a nitrogen-rich environment, Cu3SL (Figure 2). This cluster bears strong resemblance to the copper-sulfide cluster, CuZ, in nitrous oxide reductase, which activates N2O (a potent greenhouse gas) to liberate N2 and water. Our synthetic analogue is readily reduced by one electron, and further reactivity studies of this cluster are in progress. In addition, Cu3(N2)L reacts directly with N2O and other O-atom sources to generate an oxo-bridged cluster, Cu3OL, with Se to afford the selenide-bridged tricopper compound, or with N-atom sources (e.g.,azide) to yield the anionic nitride-bridged tricopper compound. Reaction of Cu3(N2)L with O2 in the presence of hydrocarbon substrates (e.g., toluene, ethylbenzene) results in O-atom insertion from O2 into substrate C–H bonds. Similar reactions of our tricopper(I) cluster with N-atom sources (e.g., azide) leads to the formation of a complex consistent with a nitride-bridged cluster. Reactivity studies and further characterization of these compounds is currently ongoing.
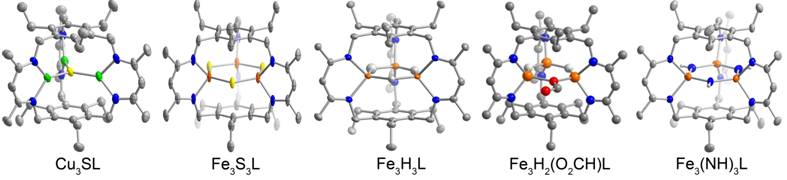
Figure 2.
Reaction of Fe3Br3L with KEt3BH or Ph3CS- affords the trihydride complex, Fe3H3L, or the triferric tri(sulfide), Fe3S3L, clusters. The hydride structure contains unusually long Fe–H bond distances and acute N–Fe–N bite angles, which highlights how the structural constraints imposed by the ligand can perturb the cluster geometry. Metal hydrides are important species in industrial and biological catalysis, and these compounds provide an opportunity to understand the reactivity of hydride clusters in a rational manner. The sulfide-bridged compound has an unprecedented planar arrangement of the metal and chalcogenide ions. This geometry means that we can examine the effect of cluster geometry (as compared to cubane-type clusters) on redox properties and reactivity. Metal-sulfide clusters are prevalent cluster in biology as well as being important materials for light harvesting (e.g., MoS2) and industrial catalysis. Fe3H3L reacts selectively with CO2 to give Fe3H2(OOCH)L (Figure 2), but the trihydride is unreactive towards all other substrates tested (inc., CH3CN, CS2, C2H2) at room temperature. This result is unusual and sharply contrasts the reactivity of the mononuclear hydride complexes. The corresponding tricobalt and trizinc tri(hydride) complexes also demonstrate similar selectivity for CO2, suggesting that steric effects may dominate reactivity. Reduction of Fe3Br3L with 6 equiv. of KC8 under an N2 atmosphere results in cleavage of the N=N as evidenced by reactions using isotopically-enriched 15N2, the release of ammonia upon acid decomposition of the product, and the solid-state structure of the isolated product (Figure 2). From both THF and toluene reactions, the isolated product Fe3(NH)3L contains three protonated N-atom bridges, which suggests that intercomplex cooperativity and X–H bond activation (X = C or O) or protonation likely occur. This complex is the first designed metal cluster competent for N2 activation.
Finally, we have demonstrated the selective synthesis of dinuclear clusters with proximal hydrogen bonding interactions by partial metalation of our hexacarboxamide cryptand with either nickel(II) or copper(II) (Figure 3). The elaborate hydrogen bonding network within the internal cavity of ligand is reminiscent of secondary coordination spheres in enzyme active sites. We have observed that the di(hydroxonickel) complex reacts rapidly and irreversibly with CO2 from air to generate a bicarbonate complex. Our inability to displace CO2 either under vacuum or upon sparging differs from comparable monometallic compounds and suggests a similar strategy can be applied to design high affinity CO2 sequestration materials.
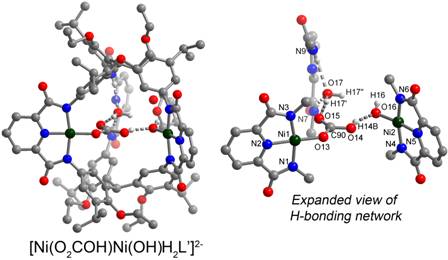
Figure 3.










