AmericanChemicalSociety.com
Reports: AC1 47257-AC1: Methods for Ligand Scaffold Optimization in Catalysis
This grant seeks to develop and exploit ligand scaffold optimization as a novel strategy for improving catalyst efficiency. The overall goal of this proposal is to develop methodologies which allow for scaffold optimization and use it to design, prepare, and evaluate focused libraries of new chiral ligands for use in catalytic asymmetric synthesis. The specific aims are to (i) carry out mechanistic studies to improve our understanding of the current generation of self-assembled ligands (SALs), (ii) evaluate new, complementary structural motifs and metal coordination geometries for the preparation self-assembled ligands and catalyst systems, (iii) evaluate the use of boracycles as an alternative motif for self-assembled ligands, and (iv) generate diverse ligand scaffold libraries via click chemistry.
Progress over the past year has been made on several of the specific aims, particularly, specific aim i. Hydrogen-bonding and metal complexation are common structural motifs used in Nature to direct the three dimensional assembly of a macromolecular framework in order to create the chiral catalytic pocket and position the functionalities involved in biological catalysts. We are exploring the latter of these strategies to organize chirality and define the topography for supramolecular catalysts. While significant progress has been toward understanding how to optimize the performance of chiral catalyst using classical approaches, much less is known on how to optimize the performance of supramolecular catalysts. The hetereobimetallic complex, illustrated schematically in Figure 1, defines a series of self-assembled supramolecular catalysts. These heterobimetallics incorporate one metal, Zn(II), whose key role is structural, and a second metal, designated Mcat, whose role is to catalyze the desired reaction. The chirality-directed self-assembly of bifunctional subunits with Zn(II) is exploited to create a library of self-assembled ligands (SALs) with distinctive scaffolds. These are combined with a second metal (e.g., Rh(I)) whose role is to promote the desired chemical reaction and complete the supramolecular catalyst.
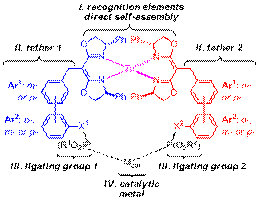
Figure 1. The general structure shown above represents a series of heterobimetallic supramolecular catalysts derived from self-assembled ligands (SAL(SR)) in which combinations of tethers 1 and 2 afford a collection of catalyst scaffolds.
These supramolecular catalysts are modular in design and comprised of four subunits. The chiral bisoxazolines (box) serve as the recognition elements (subunit I) to direct self-assembly via chiral discrimination. Each box subunit bears a variable linker or tether (subunit II) consisting of an aryl or biaryl ring whose structure is further varied by the ortho-, meta- or para-substitution pattern of those rings. Each tether subunit is terminated by a hydroxyl (tethers A-H) or hydroxymethyl (tethers I-P) group. The hydroxyl functionality serves as the attachment point for the ligating group (subunit III). Combining (S,S)- with (R,R)-box derivatives bearing chiral phosphite terminated tethers (i.e., the combination of subunits I-III) permits the facile preparation of hundreds of chiral bidentate self-assembled ligands, SAL(SR). The metal ultimately responsible for catalysis is subunit IV; it in combination with the SALs affords a series of heterobimetallic supramolecular catalysts, each possessing a slightly different catalyst scaffold.
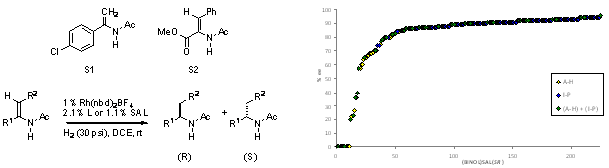
Figure 2. The results obtained in the asymmetric hydrogenation of two prototypical enamide substrates sorted from low to high as a function of catalyst scaffold (BINOL)SAL(SR). The yellow data points incorporate only tethers C-G, the blue only I-P, and the green are mixed combinations of (C-G) with (I-P).
In the present study of supramolecular catalysts for asymmetric hydrogentation, the ligating group is restricted to a chiral phosphite and the catalytic metal limited to Rh(I). The rhodium-catalyzed asymmetric hydrogenations of two prototypical substrates, S1 and S2, were examined using in situ generated SALs in combination with Rh(nbd)2BF4 at the 1% catalyst loading level. We find that subtle changes in the catalyst scaffold affect the flexibility and conformation of the catalytic pocket of supramolecular catalysts affording dramatically different selectivity and reactivity in the asymmetric hydrogenation of several prototypical enamides (Figure 2). Analysis the data reveals that each subunit within the catalyst exerts considerable influence on the outcome of the reaction. For example, the results obtained with best performance catalyst and five close structural analogs show the effect of small changes in the catalyst scaffold (Figure 3)
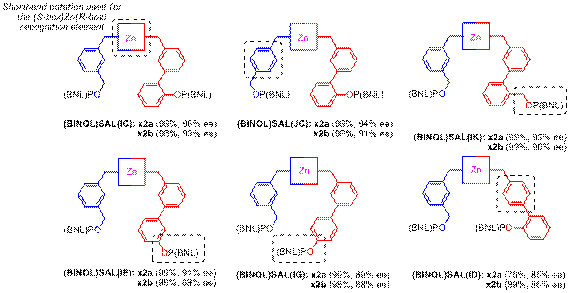
Figure 3. The results obtained with the best performance catalyst and five analogs. (Note that the structure of subunit I is represented by a shorthand notation.)
Overall, the supramolecular catalysts defined above demonstrate the potential to fine tune catalysts in ways not available with the traditional approaches. Traditional approaches are often characterized by attempts to make small structural changes in the ligands that result in large, unpredicitable changes in catalyst performance. The difficulty in fine tuning is perhaps due to the reliance on small, rigid chelates or non-bonded interactions in traditional approaches. In the present study, the formation of macrocyclic chelates, wherein small changes in structure often translate to incremental change in performance. This makes it possible to achieve a relatively smooth variation in catalyst performance over a wide range. The results demonstrate the ways in which subunits I-III influence the efficiency of the resulting supramolecular catalyst. A manuscript describing the completed study is in preparation.
Two other manuscripts are also in preparation. A one year no cost extension of the grant is requested to complete this project.
