AmericanChemicalSociety.com
Reports: B3 47113-B3: Electrochemical Activation of Cytochrome P450
During the third funding period of our project we continued our efforts to reductively activate the heme domain of cytochrome P450 from Bacillus megaterium (hBM3) using electrochemical methods. Our previous work focused on labeling hBM3 for electrode attachment by engineering a series of single surface-cysteine mutants followed by treatment with the thiol-specific N-(1-pyrene)-iodoacetamide reagent. Incubation of the resulting conjugates onto graphite electrodes gave robust protein films whose ET rates for Fe(III) reduction spanned almost three orders of magnitude. Each of the films efficiently catalyzed O2 reduction, and the product distribution (peroxide or water) correlated tightly with the rate of ferric heme reduction: ET rates greater than ~ 10 s-1 gave water exclusively, while rates less than ~1 s-1 resulted in peroxide. Despite the ability to generate ferrous heme electrochemically, none of these systems supported substrate catalysis.
One element common to all of these films is that the heme domains were wired into an electrode surface which supplied essentially an unlimited flow of electrons. This situation contrasts the native system in which two tightly regulated one-electron transfer events leads to formation of compound I. Thus during the past year we initiated studies to incorporate an internal ET relay into these conjugates (akin to the external reductases of the native enzyme) designed to deliver two electrons in discrete one-electron steps.
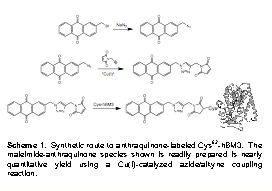
| Scheme 1. Synthetic route to anthraquinone-labeled Cys62-hBM3. The maleimide-anthraquinone species shown is readily prepared is nearly quantitative yield using a Cu(I)-catalyzed azide/alkyne coupling reaction. |
 As an initial
target, we prepared the mono-functionalized anthraquinone shown in Scheme 1.
Bromomethyl anthraquinone is first converted to the corresponding azide,
and is subsequently liked to a maliamide/ethynyl species (prepared in two steps
from propargyl amine and maleic acid) via Cu(I)-catalyzed
azide/ethynyl coupling. This reagent readily labels the surface cysteine
of mutant hBM3 through the maleimide moity. The resulting protein complex binds
to glassy carbon, (presumably through the anthraquinone) and exhibits a 1e-
heme FeIII/II couple followed by 2e- reduction of the
quinone at more negative potentials.
As an initial
target, we prepared the mono-functionalized anthraquinone shown in Scheme 1.
Bromomethyl anthraquinone is first converted to the corresponding azide,
and is subsequently liked to a maliamide/ethynyl species (prepared in two steps
from propargyl amine and maleic acid) via Cu(I)-catalyzed
azide/ethynyl coupling. This reagent readily labels the surface cysteine
of mutant hBM3 through the maleimide moity. The resulting protein complex binds
to glassy carbon, (presumably through the anthraquinone) and exhibits a 1e-
heme FeIII/II couple followed by 2e- reduction of the
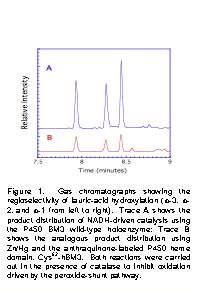
quinone at more negative potentials. | Figure 1. Gas chromatographs showing the regioselectivity of lauric-acid hydroxylation (w-3, w-2, and w-1 from left to right). Trace A shows the product distribution of NADH-driven catalysis using the P450 BM3 wild-type holoenzyme; Trace B shows the analogous product distribution using Zn/Hg and the anthraquinone-labeled P450 heme domain, Cys62-hBM3. Both reactions were carried out in the presence of catalase to inhibit oxidation driven by the peroxide-shunt pathway.
|
Notably, Introducing dioxygen causes a catalytic reduction current upon generating FeII (similar to the Py-labeled system) but the 2e- quinone couple remains nearly reversible. It is well known that doubly reduced quinones reduce dioxygen to peroxide in aqueous solution--an undesirable side reaction in our system. However, the reversible electrochemical response of the quinone in the presence of dioxygen demonstrates that the rate of the quinone/dioxygen side reaction is slow on the electrochemical timescale and not competitive with the corresponding reduced heme/dioxygen rate.
To validate our approach, we carried out substrate turnover experiments using amalgamated zinc as the heterogeneous reducing agent, and anthraquinone-labeled hBM3 conjugated at position 62. The pyrene-labeled version of this mutant catalyzed the 2e- reduction of dioxygen in the electrochemical studies described above. As illustrated in Figure 1, this system catalyzed the oxidation of the long-chain fatty acid lauric acid with a product distribution identical to that of the NADH-driven wild-type holoenzyme. Importantly, the presence of catalase has no effect on substrate oxidation: this result eliminates the peroxide shunt as the catalytic pathway. Because Zn/Hg does not reduce unlabeled ferric hBM3, we presume that enzyme activation proceeds via heterogeneous reduction of the anthraquinone followed by ET into the heme center from the quinone "reductase". Clearly, an extremely high priority for our future research will be to characterize this reaction (turnover numbers, rates) and to investigate other substrates and BM3 mutants.
