AmericanChemicalSociety.com
Reports: AC3 48295-AC3: Novel Water-Soluble N-Heterocyclic Carbene Chelates for Transition Metal Catalysts
Our efforts to design new selective hydrogenation catalysts include three approaches: 1) use of tris(triazolyl)borate (Ttz) and bis(triazolyl)borate (Btz) ligands, 2) use of chelating bis N-heterocyclic carbene (NHC) ligands, and 3) design of new ligands that provide hydrogen bonds near the metal center. Our initial efforts have focused on the use of ruthenium for ease of synthesis and stabilization of hydride complexes, but in the long term we are still interested in replacing Ru with Fe, a more readily available metal. We have begun looking at transfer hydrogenation using isopropanol as the H2 source, but in the long term hydrogenations under H2 gas allow for a more atom economical transfer of H+ and H-.
1) Use of tris(triazolyl)borate (Ttz) and bis(triazolyl)borate (Btz) ligands
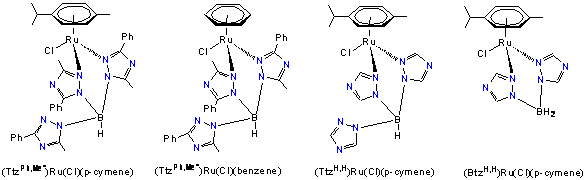
Complexes of the type (arene)RuCl(N,N) [where (N,N) is chelating tridentate ligand Ttz or the bidentate ligand Btz] are efficient catalysts for the transfer hydrogenation of ketones in the presence of isopropanol and base co-catalyst. In these complexes, tridentate Ttz ligands coordinate to the metal in a bidentate fashion with one dangling triazole ring not coordinated to the metal center. The base co-catalyst (KOH) likely generates a hydride species that can serve as the active catalyst; in related experiments we have been able to observe formation of a hydride complexes.
In order to investigate the role of sterics and the uncoordinated triazole ring, we have made a series of new complexes [Chart 1], (TtzPh,Me*)Ru(Cl)(p-cymene), (TtzPh,Me*)Ru(Cl)(benzene), (TtzH,H)Ru(Cl)(p-cymene), and (BtzH,H)Ru(Cl)(p-cymene) [the superscripts indicate the alkyl substituents on the ring, * indicates a rearranged ligand]. These were characterized by spectroscopic, analytical and X-ray diffraction techniques. These are air stable complexes and can be handled in air and in the presence of moisture, a feature convenient for green chemistry applications. All four compounds catalyze transfer hydrogenation of several aromatic ketones in the presence or absence of base (KOH). In the absence of KOH, hydrogenation proceeded slowly and took ~5 days to achieve >90% conversion. However, the presence of base accelerated the hydrogenation and >90% conversion is observed in 24 h [Reaction conditions: 2.0 mmol ketone, 2.0 mmol base (KOH, 0.2 M, 10 mL in i-PrOH), catalyst 1.0 mole%, 24h reflux]. To measure the selectivity of hydrogenation of polar bonds (C=O) over C=C, we also performed the hydrogenation of Ph-CH=CH-C(=O)CH3 using the catalysts mentioned above. A mixture of products indicating both C=C (alkene) and C=O hydrogenation is observed, however it appears that the reaction is more selective for C=O at earlier reaction times.
Chart-1
We
are also interested in transfer hydrogenation of ketones
using formate and formic acid as the H2
source. This reaction liberates CO2(g), and the reverse reaction is of great
interest as the first step in the conversion of CO2 to liquid fuels
like methanol. The hydrogenation of acetophenone in the presence of HCOOH/HCO2Na (5 equivalents of each relative to acetophenone) in water with (TtzPh,Me*)Ru(Cl)(p-cymene) as a catalyst (1
mol%) showed ~40% conversion to hydrogenation products, which is a promising
initial result. 2)
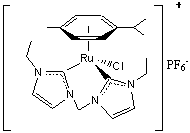
Use of chelating bis N-heterocyclic carbene (NHC) ligands A ruthenium (II) complex supported by NHC
ligands (Fig. 1) was used as a hydrogenation catalyst
with ketone substrates. This complex has been
characterized by 1H NMR and X-ray crystallography. Typically, yields >90% were achieved for
the reduction of ketones with 1 mole % catalyst, 100%
KOH, and isopropanol as the hydrogen source. The
catalyst seems to have good functional group tolerance as high percent
conversions were achieved in the presence of other functional groups and/or heteroatoms (N, Br, OMe).
With H2 gas (15 psi) as the hydrogen source, acetonitrile
as solvent, 1 mole % catalyst, and 100% KOH, 8.00 % conversion of acetophenone to 1-phenylethanol was observed after 24 hours.
The low percent conversion is most likely due to the low pressure of hydrogen
gas, but future experiments will be done at higher pressures now that we have
equipment that can handle pressures up to 100 psi.
Figure
1 Mechanistic studies in the
literature for similar transfer hydrogenation reactions suggest that the
chloride ligand in precatalysts
is replaced by a hydride ligand to serve as the
hydride source. Efforts to isolate and
observe a ruthenium hydride complex synthesized from the complex in Fig. 1 have
been underway. Upon treatment with NaOMe and methanol, a hydride ligand
was observed in the 1H NMR at -10.3 ppm at
33% conversion. When using KOH and methanol a hydride ligand
was also observed at -10.3 ppm but in small quantity. 3)
Design of new ligands that provide hydrogen bonds
near the metal center We have also
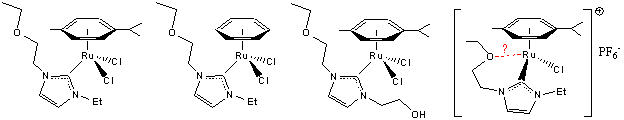
been interested in the development of new NHC ruthenium complexes in which
dangling ether and/or alcohol groups are present [Chart 2]. Ether groups can
accept hydrogen bonds and donate them when protonated. Preliminary spectroscopic and/or
crystallographic data support the structures shown and further studies are in
progress. Efforts to synthesize tris(pyrazolyl)borate ligands with
pendant amino groups are in progress. We
have made tris(3-amino-t-butyl-5-methyl-pyrazolyl)borate which will
provide an NH group for hydrogen bonding to ligands
(like hydride) that are on the metal center.
Other groups have shown that pendant hydrogen bonding groups can greatly
improve rates of catalytic hydrogenation. Chart-2
Impact
on the Career of the PI and the Students The funding from PRF has allowed the Papish group to gain preliminary results for a funding
application to DOE under the Early Career Program; the PI's pre-application was
successful and a full application is in being written. Several students and postdocs
have worked on this project and used it to further their career
aspirations. Postdoc
Mukesh Kumar is currently seeking academic jobs and Ismael Nieto will be
seeking a job in the coming months; for both the PRF project has been valuable
research experience. Joseph DePasquale is currently a third year graduate student, and
this project will form the beginning of his thesis. Undergraduate Stephen Fredericks has worked
on this project and his experience will help in his graduate school
applications. Soon we hope to submit two
papers from the group's work on the above project.