AmericanChemicalSociety.com
Reports: AC2 46636-AC2: Quantum Mechanical Calculation of Hydrogen Isotope Exchange Thermodynamics and Kinetics on Petroleum Model Compounds
The third year of research into the thermodynamics and kinetics of H-D isotopic exchange on kerogen-related molecules was conducted by performing calculations on H abstraction and H-D exchange reactions with hydroxyl groups representative of those on mineral surfaces (e.g., clays). As kerogen interacts with the minerals of the host rock or as petroleum migrates through sedimentary rock, H-D exchange may occur. This research will be used to help interpret observed H-isotope fractionation factors in ancient organic matter from sedimentary rocks. Theoretical values are needed because molecular-specific isotopic analyses have recently been made possible, but experimental values are difficult to obtain especially for specific sites within compounds. Observed fractionation trends will require knowledge of both the equilbrium fractionation factors and exchange rates as a function of temperature. Knowledge of isotopic H-D fractionation factors between kerogen and petroleum components and mineral surfaces is limited.
The B3LYP/6-311++G(d,p) method was used to calculate vibrational frequencies of the organic compound 2-methyl butane (Fig. 1) and molecular clusters representive of mineral surface groups. The calculated frequencies were used to estimate ln(b) values for alkane compounds. The α values were then calculated by the difference between the alkane ln(b) values. This method of calculating ln(b) values has been previously tested by comparing the model ln(β) value against the known value for water vapor.
In general, the results (Table 1) predict that D prefers the aluminol (Al-OH) and silanol (Si-OH) groups over the C-H bond in 2-methyl butane. Exceptions to this are the secondary and tertiary C-H groups exchanging with Al-OH and SiOH and 25°C. At 100°C, these C-H groups slightly prefer D over the Si-OH sites but not the Al-OH sites. Al-OH2 sites (i.e., Al-OH group protonated at lower pH) are predicted to fractionate in favor of D at both temperatures.
Table 1 – Calculated α and β values at 25 and 100°C using B3LYP/6-311++G(d,p). The “Atom” column is the C atom type in 2-methyl butane. The sites for exchange on 2-methyl butane are labeled in Fig. 1. “Al-OH”, “Al-OH2” and SiOH represent hydroxyl exchange sites on aluminosilicate minerals such as clays.
Atom β αAl-OH αAl-OH2 αSiOH____
25°C
Alpha 11.412 0.903 0.822 0.971
Beta 11.585 0.917 0.834 0.986
Secondary 12.653 1.002 0.911 1.077
Tertiary 13.296 1.053 0.957 1.132
100°C
Alpha 6.104 0.902 0.838 0.940
Beta 6.174 0.913 0.847 0.950
Secondary 6.523 0.964 0.895 1.004
Tertiary 6.726 0.994 0.923 1.035
Table 2: Calculated activation barrier energies (in kJ/mol) for H abstraction from each 2-methyl butane H site with each type of surface hydroxyl (see Fig. 2). The activation barrier energies are calculated from the energy-minimized structure of 2-methyl butane and the O of the surface. The bonds were brought together in 0.2 Å increments up to a distance of 1.05 Å for the O-H bond. Two sites for the Si surface were examined: SiO- and SiOH.
Molecule Site Al-OH Al-H2O SiO- SiOH
2-Methyl Butane Alpha 148 184 185 287
Beta 140 180 193 282
Secondary 155 160 195 295
Tertiary 168 175 188 290
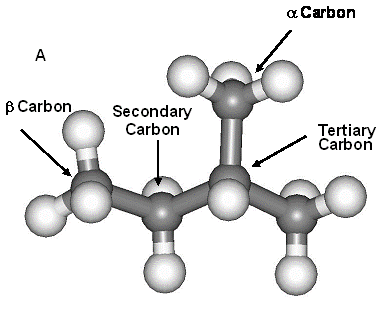 Figure 1: The molecule structures that
were modeled a) 2-methyl butane and b) 2,6-octane.
The four separate H positions are labeled in each molecule – tertiary,
secondary and two primary H atoms (one a
and one b to the tertiary C atom).
Figure 1: The molecule structures that
were modeled a) 2-methyl butane and b) 2,6-octane.
The four separate H positions are labeled in each molecule – tertiary,
secondary and two primary H atoms (one a
and one b to the tertiary C atom).
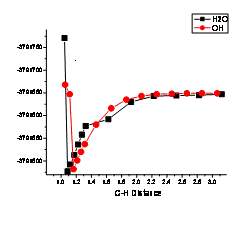
Figure 2: H abstraction between the reaction pathway between a model Al2(H2O)4(OH)6 surface and the secondary carbon site on 2-methyl butane. A H2O site on the Al surface, deprotonates after 2-methyl butane deprotonates, thus 2-methyl butane accepts the H from the Al-OH2 site.
The most important conclusion of this study of organic-mineral H-D isotope exchange is seen in Table 2. The calculated activation energies are in the range of 140 to 300 kJ/mol. Compared to calculated H-D exchange activation energies for 2-methyl butane and H2O (in the range of 200 to 300 kJ/mol), these can be dramatically lower for all C-H sites when interacting with surface aluminol groups. Consequently, we conclude that H isotope exchange can occur at significantly lower temperatures and with faster rates when kerogen and petroleum interact with aluminosilicate mineral surfaces. Because the surface hydroxyl H isotope signature is determined by interactions with pore waters, this ends up being a way to catalyze H isotope exchange between water and organic matter at depth – either in the source rock or durnig migration of fluids.
