AmericanChemicalSociety.com
Reports: UNI1 49471-UNI1: Aziridination and Retro-Aldol Fragmentation of Dioxenones: Application in the Synthesis of α-Amino Acids
Project Overview
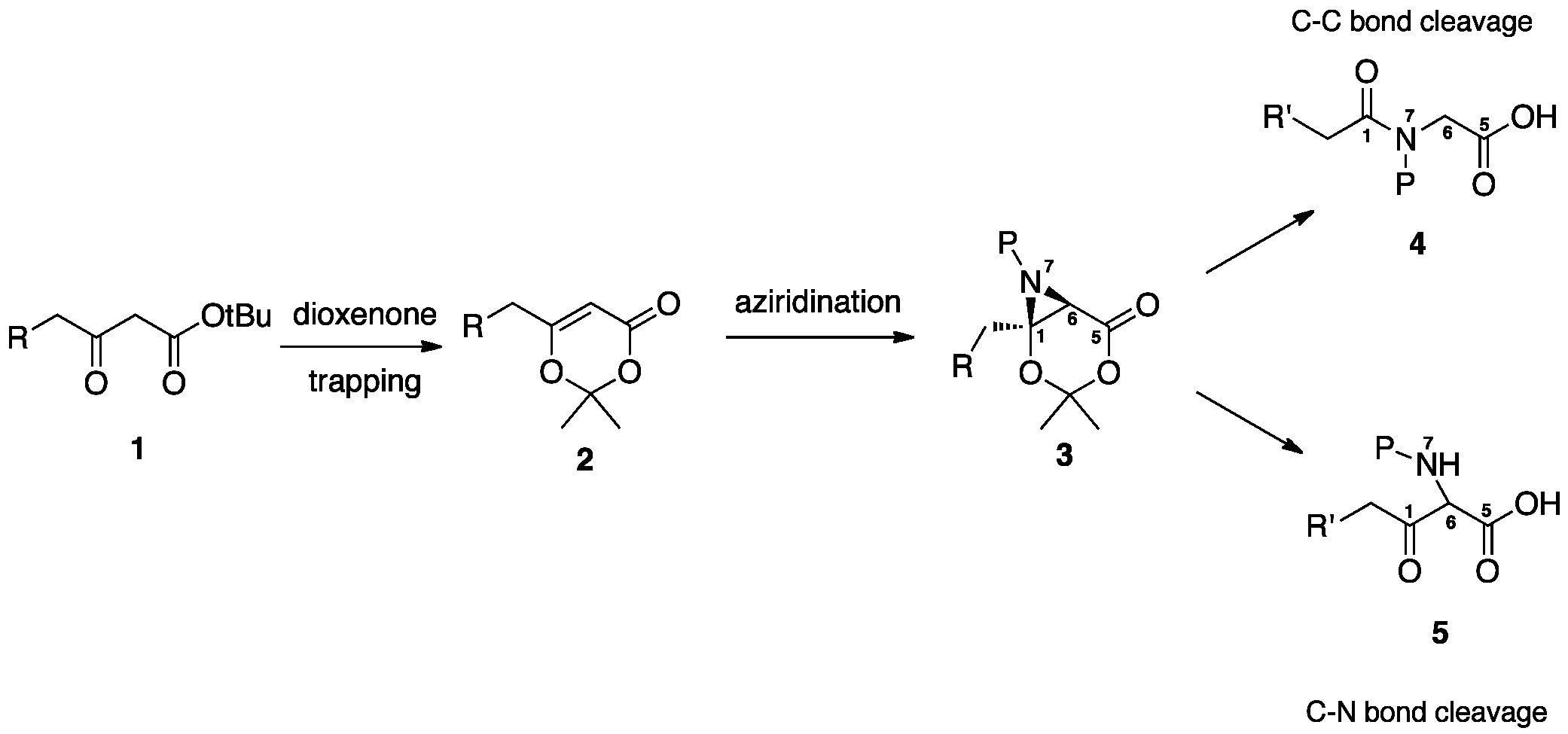
The nucleophilic ring opening of aziridine carboxylates has been a well-studied method in the preparation of non-naturally occurring amino acids. However, the routes to the preparation of these aziridines vary widely and are somewhat limited in substitution and complexity. One effective method for incorporating nitrogen into organic molecules with varying complexity is by the direct aziridination of alkenes. If the direct aziridination of variably substituted olefins can be combined with a general aziridine ring opening protocol, rapid access to an assorted collection of non-proteinogenic amino acids can be realized. The method that we are investigating in the laboratory involves the simple construction of a variably substituted b-ketoester, such as 1, that is covalently trapped in its enolic form as a 1,3-dioxen-4-one 2 (Scheme 1). The development of an aziridination protocol for the dioxenones, then investigation of the retro-aldol fragmentation of the resultant bicyclic structure will furnish amino acids and amides of variable substitution.
Scheme 1
Despite a whole
host of examples of aziridines in synthetic chemistry, there are only a few
reports of aziridination of enol ethers and a,b-unsaturated
esters and even fewer examples involving both the enol and ester
functionalities in the same molecule, such as the 1,3-dioxen-4-one with which
we are working. Most commonly,
aziridines are formed by the generation and transfer of a high-energy nitrene
to an available alkene. The
generation of nitrenes can be achieved by photolysis or thermolysis of an
azide, or else the formation of a metal-nitrene from iminoiodinanes, among
other methods. Our work
surrounding the direct aziridination of dioxenone substrates during the funding
period of September 2009 to August 2010 is summarized below.
Summary
of Experimental Work:
Aziridination of 1,3-Dioxen-4-ones The direct
aziridination by metal nitrene insertion for 2,2,6-trimethyl-1,3-dioxen-4-one 6
to afford compound 8 is the first method we have investigated with very
little success (Scheme 2). We
prepared three different aryl iminoiodinanes, utilized a sampling of copper and
dirhodium metal catalysts, and also varied solvent and temperature conditions
yet we never observed the formation of 8. We quickly
abandoned that route and turned our efforts toward the both the Michael
initiated ring closure using ethyl nosyloxycarbamate and the metal mediated
oxidation of N-aminophthalimide (Scheme 2). Again, our successes with these methods
were limited. In the latter
pathway, we were able to observe product 9
as a mixture in 1HNMR and as evidenced through LC/MS analysis.
Scheme 2
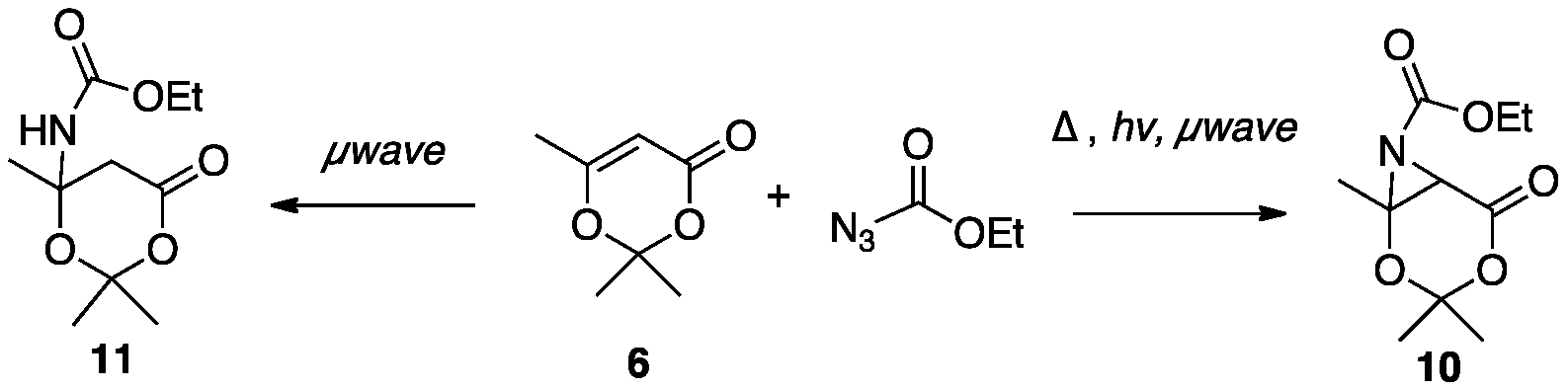
Over the last
nine months, we have focused most of our attention on employing ethyl
azidoformate as a nitrene precursor to affect our desired aziridination. With precedent existing for nitrene
formation under photolytic or thermolytic conditions, we used this as a jumping-off
point (Scheme 3). We were able to
design an NMR study for the irradiation of 6 that mirrored the thermolysis.
In each case, the bicyclic product 10 was found by GC/MS in less than 30 % conversion and
became an almost impossible feat to isolate. The thermal stability of the dioxenone starting material as
well as the resulting product has now come into question. Scheme 3
With the
acquisition of a new microwave reactor we envisioned that utilizing microwave
irradiation for the synthesis of thermally sensitive products such as 10, we hope to be able to improve the yields of these
reactions through lower temperature, milder conditions, and shorter reaction
times. This summer we were able to almost cleanly identify the two major
products in the aziridination of 6
with ethylazidoformate. We now
have 1H NMR and GC/MS data that supports the formation of both
aziridine 10 and Michael-type
adduct 11. The details of this work were presented
at the National American Chemical Society Meeting on August 22nd,
2010 in Boston, MA with two Bard undergraduates – Madison Fletcher '12
and JosŽ Luis Falla '13. We are in
the process of reproducing our data, generating parallel examples, preparing
and testing variably substituted dioxenones, and determining by what
mechanistic pathway the unexpected product 11 is afforded.