AmericanChemicalSociety.com
Reports: AC4 47590-AC4: Design of New Radical Reactions: From Elusive 5-Endo-Dig Cyclization to Cascade Transformations
New Radical Cyclizations: We, for the first time, offered
theoretical analysis of a scarcely studied radical cyclization – the 5-endo-dig
process. Although this path is apparently favorable according to the In the second theoretical development, we analyzed the
competition between two important radical cyclization processes – 5-exo-dig and
6-endo dig cyclizations (JACS, 2005, 12583).
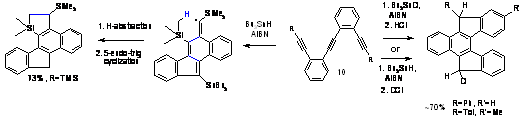
These processes are used in organic synthesis and are implicated in the process
of formation of polycyclic aromatic compounds and carbon nanostructures. When the acetylene moiety and the radical are
connected through a saturated two-atom bridge, the 5-exo process is strongly
favored kinetically. However, when the bridge is unsaturated, the 6-endo
products are stabilized by aromaticity. Part of the product stabilization is
transferred to the decrease in the 6-endo activation barrier rendering it
kinetically competitive with the 5-exo-dig path. As a result, the 5-exo or 6-endo selectivity
can be fine-tuned by seemingly subtle modifications in the structure of the
starting materials. Our study determined the contributions of thermodynamic and
strain effects to the 5-exo/6-endo competition, extended these findings to
available experimental data and provided predictions to guide our experimental
studies which led to the development of an efficient radical cascade providing polyclic systems representing the tip part of carbon nanotubes (JACS, 2008, 11535).
Ortho-effect in the Bergman cyclization: kinetics and development of
new radical cascades: We applied steric and
electronic effects of ortho substituents for efficient control of thermal
Bergman cyclizations of benzannelated
enediynes. Theory predicts that change in ortho
substituents can lead to a nearly 2000-fold difference in the cyclization rate
(250-fold greater range than for para substituents).
We confirmed these computational
predictions with experiments and proved that steric
and electronic effects of ortho substituents can be used for efficient control
of thermal Bergman cyclizations of benzannelated enediynes.
Interception of p-benzynes by intramolecular H-abstraction increases the apparent
reaction rate by rendering the cyclization step effectively irreversible. From
a chemical perspective, such interception is interesting because it produces a
new, more persistent diradical which is not capable
of deactivation through the retro-Bergman opening. In addition, intramolecular H-abstraction in OMe-substituted
p-benzynes
followed by domino radical cyclizations can be used
to transpose radical centers in p-benzyne and create
new types of diradical species. Interestingly, the same step is possible in
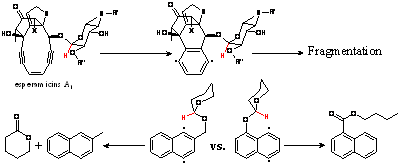
natural enediyne antiobiotics
of calicheamicin and esperamicin
families where a OCHR substituent is similarly
positioned relative to the p-benzyne
intermediate. We found that such hydrogen abstraction can rationalize the so
far unexplained fragmentation of esperamycin A upon
its activation towards cycloaromatization. In
addition, an interesting radical rearrangement which transposes oxygen and
carbon atoms attached to an aromatic ring was discovered experimentally (JACS, 2010, 133,
967-979).
Anionic cyclizations
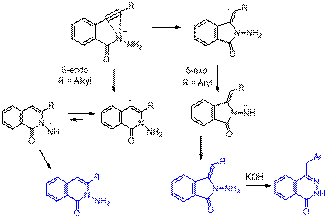
of alkynes: We have also combined theory
and experiment to analyze the 5-exo/6-endo selectivity in digonal cyclizations of N-nucleophiles (J. Org. Chem. 2009, 74
, 8106–8117). We can fine-tune the regioselectivity for nucleophilic closures via modulation of electronic
properties of the alkyne moiety. Alkyl substituents at the alkyne
terminus favor the 6-endo-dig closure whereas aryl groups greatly facilitate
the alternative 5-exo-dig path.
Competing cyclization
pathways were fully analyzed computationally. Decreased 5-exo-dig activation
energies and increased stability of the 5-exo products for R = Ph confirm that
the Ph group steers the cyclization selectively down
the 5-exo path by providing benzylic stabilization to
the anionic center in the product. In
contrast, the competition between the 5-exo and 6-endo-dig closures is close
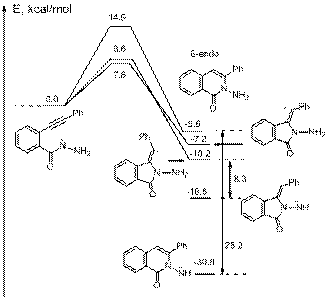
for alkyl substituted acetylenes. For R=Me, the values of cyclization
barriers are within 1 kcal/mol from each other and thus, both cyclizations can proceed with comparable rates. Although
the 5-exo cyclization has 0.6 kcal/mol lower barrier
than the 6-endo closure in the gas phase, introduction of solvation
reverses this preference. Moreover, the 5-exo-dig cyclization
is predicted to be endothermic and readily reversible in the latter, whereas
the 6-endo-dig closure is ~10 kcal/mole exothermic. The higher computed activation
barriers for both 6-endo and 5-exo cyclizations of
alkyl substituted alkynes are consistent with experimental observations. Full potential energy surface for the
competing 6-endo and 5-exo cyclizations of hydrazide anions at B3LYP/6-31+G(d,p) level of theory is given below.
These anionic cyclizations
lack a significant thermodynamic driving force because the gain in stability
due transformation of a weak p-bond
into a stronger s-bond is offset by the
transformation of a stable nitrogen anion into an inherently less stable carbanionic center. Formation of the final products is
negotiated through several proton shifts, ultimately leading to the most stable
tautomeric anion as a thermodynamic sink.. Such tautomerizations are
likely to play the key role in driving such cyclizations
to completion but may also prevent future applications of such processes as the
first step in domino cyclization processes.