AmericanChemicalSociety.com
Reports: UR4 49447-UR4: Investigating Proton-Coupled Electron Transfer with Radical Cations Appended with Bases
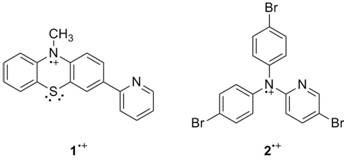
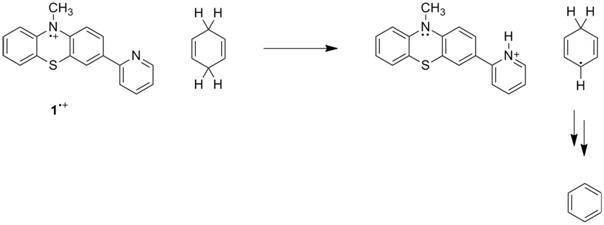
Progress in this year of the grant has focused on synthesis and characterization of radical cations 1•+and 2•+ and initial studies of their reactivity with hydrogen atom donors. Several sources in the chemical literature state that heterocyclic nitrogen radical cations rapidly decompose, as the nitrogen can act as a nucleophile. For this grant, we have synthesized one radical cation 1•+ that is stable in solution for at least an half an hour, perhaps longer. Another radical cation 2•+ appears to be stable on an electrochemical timescale, and we have made progress toward its synthesis. Cyclic voltammetry, reactivity studies and computational studies preliminarily suggest that these agents can act as hydrogen atom abstraction agents with a concerted proton-electron transfer (CPET) mechanism. These systems parallel important biological systems such as flavins and DNA and provide a better understanding of nitrogen radical chemistry. Such compounds may have use in hydrocarbon functionalization and in artificial photosynthesis.
Student
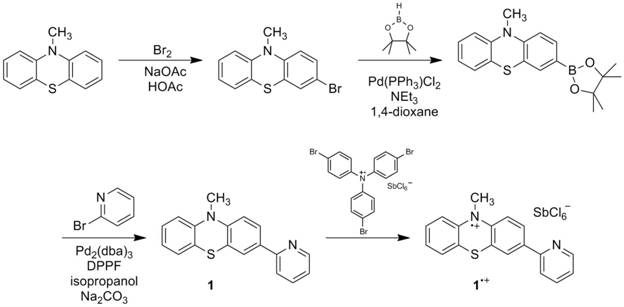
researchers Crina Sasaran, Victoria Polito and Jonathan Geruntho synthesized the
precursors for radical cations (1
and 2) using procedures directly or
modified from the literature. Cyclic
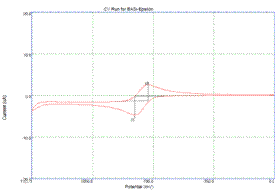
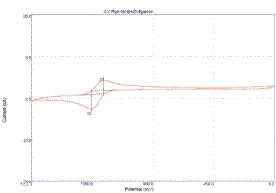
voltammetry for both compounds in 0.1 M n-Bu4NPF6 in
acetonitrile affords oxidation and reduction waves, indicating clean
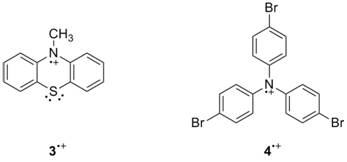
electrochemical oxidations on the timescale of the scans. The reduction potentials for 1•+/0 and 2•+/0 are 0.32 V and 0.87 V, respectively. These potentials are similar to parallel
compounds (3, 0.32 V1 and
4, 0.67 V2). The more positive potential of 2•+/0 can be explained by
the electron deficient nature of the pyridine ring. On the electrochemical timescale, the
oxidized species are stable.
Figure 1.
Synthesis of 3-(2-pyridyl)-10-methylphenothiazium radical cation.
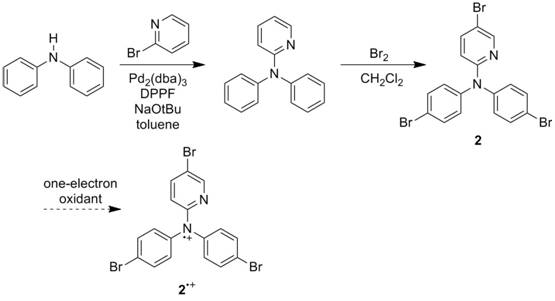
Figure 2. Synthesis of N,N-di-(4-bromophenyl)-N-(4-bromo-2'-pyridyl)aminium radical
cation.
Cyclic voltammogram of 1•+/0 Cyclic voltammogram of 2•+/0 Brittney
Tiley and Robert Richards performed one-electron oxidation reactions and
initial reactivity studies. Four
different oxidizing agents were attempted to generate 1•+ chemically: NOBF4, thianthrenium, AgPF6,
tris(4-bromophenyl)aminium hexachloroantimonate in acetonitrile or methylene
chloride. Both the silver and aminium oxidants
produced species with distinct absorptions in the visible spectrum (779 and 864
nm; further work is required to confirm the molar absorptivities) that parallel
those in 10-methylphenothiazinium radical cation (780 and 870 nm).3 In addition, these reactions gave broad 1H
NMR spectra in CD3CN. The
reduction potential for 2•+/0
indicates that only NOBF4 is oxidizing enough out of the oxidants
above to produce the radical cation from 2. Reaction led to shifted but distinct NMR
peaks and a product with an irreversible voltammogram. Likely, the NO+ ion reacts with
pyridyl rings for 1 and 2.
Two possible directions for compound 2 include reaction with SbCl5, a common oxidant for
aminium generation, or alteration of the substrate. Substitution of bromine atoms with methoxy
groups is synthetically possible. This
change will lower the reduction potential and hence allow for more possible
oxidants. Radical
cations can accept an electron, and pyridine can accept a proton. Our hypothesis is that the combination of
these two components will allow for the net transfer of a hydrogen atom by CPET. Hence, we have explored the reactivity of 1•+ and 2•+ with two hydrogen atom donors, 9,10-dihydroanthracene
(DHA) and 1,4-cyclohexadiene (CHD), in solution with UV-vis and NMR for 1•+ and with cyclic
voltammetry for both compounds.
In
preliminary cyclic voltammetry experiments with 2 and DHA in solution, the current for reduction wave is reduced
relative to that of the oxidation wave, suggesting an electrochemical-chemical
(EC) mechanism in which the chemical mechanism is bimolecular. Further studies in which DHA concentrations
and scan rates are varied and the voltammograms are modeled may be able to
afford a bimolecular rate constant for this reaction. Changes in the voltammograms with 1 and DHA or CHD in solution are less
apparent. Addition of
DHA or CHD to 2•+ in
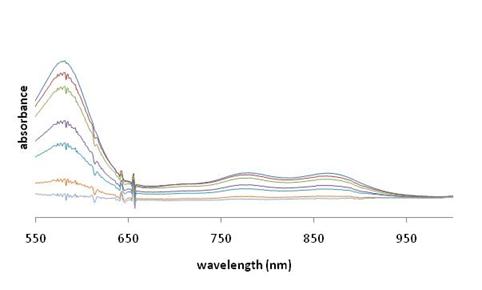
acetonitrile leads to the decay of the absorptions in the visible region of the
spectrum. In reactions with DHA and CHD,
peaks corresponding to anthracene and benzene, respectively, appear in 1H
NMR. Planned control reactions with 3•+ are required to
establish that the aromatic products are not from one-electron oxidation. Better control of quantities will establish
the stoichiometry of reaction.
Figure 3.
UV-vis scans for reaction of 2•+
and CHD. Computational
studies by Jeffrey Wolbach and Samantha Cordisco suggest that these reactions
occur with a CPET mechanism.4
The reactants and products for CPET mechanistic step for reaction
between 1•+ and CHD have
been calculated. The transition state
has been identified and verified both with imaginary frequency and intrinsic
reaction coordinate (IRC) calculations.
The barrier height is approximately 13 kcal/mol in the forward direction
and 26.5 kcal/mol in the reverse direction.
Similar calculations with DHA are under way. Some intermediates for stepwise proton transfer
or electron transfer have also been calculated and tentatively indicate that
the concerted mechanism has a much lower barrier for reaction.
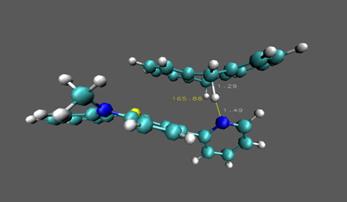
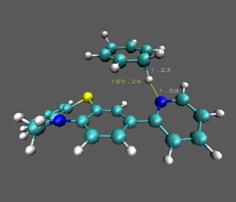
Figure 4. Calculated transition
states for 1•+ with DHA (left) and CHD (right). In the next
year of the grant, we plan to chemically generate 2•+ or a similar aminium compound and continue
reactivity studies with hydrogen atom donors for all compounds under
study. Kinetic studies will reveal the
rate law and barriers for these reactions.
Kinetic isotope effect studies will determine if the hydrogen atom is
involved in the mechanism, and computational studies will parallel the
experiments. (1) Rhile, I. J.; Markle,T. F.;
Nagao, H.; DiPasquale, A. G.; Lam, O. P.;
Lockwood, M. A.; Rotter, K.; Mayer, J. M. J Am. Chem. Soc. 2006,
128, 6075-6088. (2) Connelly, N. G.; Geiger, W. E. Chem
Rev. 1996, 96, 877–910. (3) Rosokha , S. V.; Kochi, J.
K. J. Am. Chem. Soc. 2007, 129, 3683-3697. (4) Geometries were calculated with
B972/6-31+g(d,p) with thermal corrections
with B972/6-31+g(d,p) in the gas phase.
Energies for the stationary points were calculated with
CAM-B3LYP/6-31++g(2d,p). Transition
states were calculated with ONIOM (CAM-B3LYP/6-31++g(2d,p):B972/3-21+g), and
the frequencies and IRC were calculated with ONIOM
(CAM-B3LYP/6-31++g(2d,p):B972/3-21+g).
The energies, transition states, frequencies and IRC were calculated
with implicit solvation in CH3CN.