AmericanChemicalSociety.com
Reports: AC6 47617-AC6: Examination of the Role of Entropy and Defects on the Relative Stability of Molecular Crystals
This project aims to develop and apply molecular simulation methods that can aid in the prediction of crystalline polymorphs. Prediction of stable crystalline polymorphs should be completed by examining the relative free energies of candidate structures. In practice this is almost always done by examining the lattice energy alone, ignoring entropic contributions to the free energy. The error incurred with this procedure is generally considered to be of the order of the difference between candidate structures, but nevertheless the practice persists because it is inconvenient to consider the entropy rigorously, and approximate methods that introduce entropy tend not to lead to greater success in predicting the observed polymorphs. Such applications leave open the question as to whether failure of the prediction is due to inadequacies of the molecular model, or due to the approximations introduced to estimate the free energy. A major goal of this work is to establish the significance of the entropy (and its neglect) as a source of error in polymorph prediction efforts.
Rigorous methods to compute the free energy are seldom applied because they are tedious and computationally intensive. Thus a major component of this work is the development and/or improvement of such methods. To this end we are considering techniques that can yield an accurate free energy with minimum computational effort. Granted that molecular simulation is required to render a good free-energy value, we aim to develop methods that require as few simulations as possible. Ideally one simulation would suffice, but experience with free-energy techniques tells us that at least two are needed to get an accurate and reliable result. Accordingly, the approach we are pursuing is based on a system of coupled harmonic oscillators (which if taken by itself provides a well-studied means to estimate the free energy) combined with two-stage free-energy perturbation. Appropriate two-stage methods include umbrella sampling and overlap sampling, and our studies to date have looked at the ability of these to provide accurate free energies.
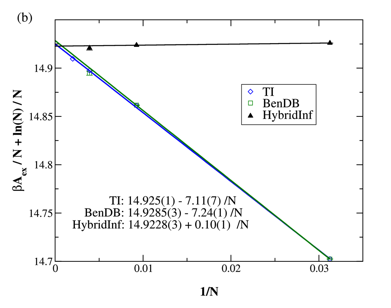
Previously we reported that such methods could be effective if applied to systems with sufficiently few molecules. The penalty for doing so is incurring finite-size effects that introduce new errors that tend to defeat the purpose of the calculation. In the time since our last report we have made an observation that circumvents this problem. In particular, we have found that the correction to the harmonic reference (the quantity that is most challenging to calculate) is virtually independent of system size. This suggests a hybrid technique in which perturbation is applied to a small system and the result added to a large-system harmonic reference to obtain a good estimate of the correct large-system free energy. The behavior is exemplified in FIG 1.
FIG. 1. Excess free energy for
soft-sphere model evaluated with various methods, as a function of the system
size used in the calculation (with infinite system a zero of abscissa) at kT/
ε = 0.5, with ρσ3 = 1.256. The notation HybridInf indicates hybrid method in
which harmonic free energy extrapolated to infinite size is used, with
correction from smaller system sizes (32, 108 and 256 particles), as plotted. Non-hybrid
alternatives are shown in other lines, and exhibit a much greater system size
dependence.
In a separate development, we have formulated a method for evaluation
of free-energy differences with temperature that is highly precise, accurate,
and efficient. We have applied the
method to evaluate free energies for soft-sphere models with unprecedented
precision. We call the method harmonically-targeted temperature perturbation
(HTTP), and involves coupling a free-energy perturbation in temperature with harmonically-based
atomic displacements that aid the perturbation calculation. The method is such
that if it were applied to a true harmonic crystal, it would yield exact
results, and consequently in practice the calculation accumulates noise only to
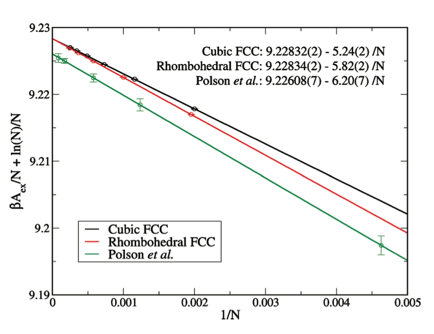
the degree that the crystal is anharmonic. The performance of the method is shown in FIG 2, which
compares results from the HTTP method to the best data available
previously. It should be noticed
both that we corrected a small error in the previous result, and we obtain free
energies with much greater precision, as indicated by comparison of the error
bars.
FIG. 2. Convergence of free energy with system size for
soft-sphere model, comparing new HTTP method with literature data of
Polson et al. [J. Chem. Phys. 112, 5339 (2000)]. HTTP
method is applied
to systems with two geometries for periodic boundaries, and agree in the
infinite-system limit (y-axis intercept).
In addition we have computed virial coefficients for the soft-sphere
models that were used to develop these simulation methods, obtaining results
that are an order of magnitude more precise than previously established, and
going one order in density higher (up to ρ8). We use the free-energy determined from the virial equation to
describe the fluid, and connect to the solid-phase free-energy results
described above to establish more accurate values for the solid-fluid
transition point (i.e., melting or freezing point). Our ultimate interest is in polymorph transitions, and we have used our
solid-phase free energy data to examine its ability to capture this behavior in
the soft-sphere model. We have taken special interest in the contributions to
the free energy from the lattice energy by itself, the harmonic contribution,
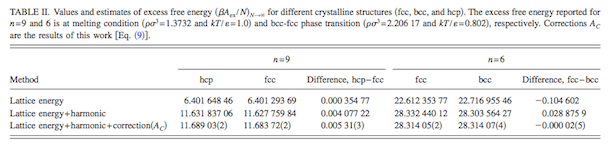
and the anharmonic correction that is the focus of this work. Data are summarized in the following
table for two soft-sphere models (indicated as n = 9 and n = 6).
The latter is known to exhibit a fcc-bcc polymorphic transition, and the table
shows that the contributions beyond the lattice energy are significant in
establishing this behavior.
The soft-sphere model is a useful starting point for these studies, but
has anomalies that make it unsuitable as a basis for general conclusions about
methods or phenomena. We are in the process of applying these methods to a
realistic model for carbon dioxide, looking in particular at its polymorphic
behavior and the relevance of entropic effects in producing and locating it. We
expect to finish that work in time for the conclusion of the grant period.