

47272-AC1
Short, Tunable Chiral Peptidic Ligands for Osmium Tetroxide-Mediated Chemistry
During the course of
our investigation of the Cinchona alkaloid mediated enantioselective chlorolactonization
protocol, we sought a general and operationally simple methodology for
preparing various N-chlorinated In
2002, Braddock and coworkers disclosed an effective catalyst for the bromolactonization
of olefinic carboxylic acids employing ortho-iodoarylamide (e.g. 14) and amidine (e.g. 15) catalysts in the presence of
N-bromosuccinimide (NBS). These
catalysts, shown to proceed via an iodine (I)/iodine (III) oxidative couple (16), showed a significant increase in
rate relative to the uncatalyzed reaction with NBS alone (Figure 3).
Subsequently, they reported that a similar rate acceleration could be
accomplished by employing non-iodinated amidines. We surmised that incorporating this general catalyst
scaffold within a chiral peptide framework might garner some selectivity in the
process. With this in mind, we
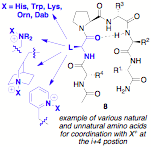
have prepared and evaluated roughly 40 peptides for the asymmetric
bromolactonization of compound 17. The
preparation of such catalysts is facilitated by Fmoc solid phase peptide
synthesis (SPPS). Importantly, we
have discovered an initial "hit" scaffold that returns the desired bromolactone
19 in 14% ee when 0.1 equivalents of peptide 18 is employed. It is likely that continued exploration
of the "chiral space" about this promising scaffold, available through natural
and unnatural amino acid building blocks will lead to an optimized
catalyst. Such combinatorial
approaches driven by directed screening about an initial "hit" scaffold is a
proven strategy for catalyst design.
 In sharp contrast to the large
number of known examples of substrate controlled stereoselective
halolactonizations, reagent controlled processes are exceptionally rare, and have only
begun to emerge within the last 15 to 20 years. The advantages of a successful approach to a reagent
controlled halolactonization are obvious, since such a methodology would
provide rapid access to richly functionalized halolactones in one step from
achiral congeners. Utilizing the
funds from PRF, we have initiated a comprehensive research program that is
charged with evaluating new asymmetric catalytic systems that are obtained
easily and can be rapidly synthesized and screened. These include peptidic scaffolds and non-peptidic motifs (at
times a mix of the latter two systems).
In particular, we will evaluate these chemistries via the OsO4
mediate oxidation of olefins and the halolactonization reactions of
hydroxyalkenes.
In sharp contrast to the large
number of known examples of substrate controlled stereoselective
halolactonizations, reagent controlled processes are exceptionally rare, and have only
begun to emerge within the last 15 to 20 years. The advantages of a successful approach to a reagent
controlled halolactonization are obvious, since such a methodology would
provide rapid access to richly functionalized halolactones in one step from
achiral congeners. Utilizing the
funds from PRF, we have initiated a comprehensive research program that is
charged with evaluating new asymmetric catalytic systems that are obtained
easily and can be rapidly synthesized and screened. These include peptidic scaffolds and non-peptidic motifs (at
times a mix of the latter two systems).
In particular, we will evaluate these chemistries via the OsO4
mediate oxidation of olefins and the halolactonization reactions of
hydroxyalkenes. 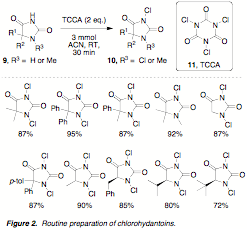 Preliminary experiments have
uncovered a viable, selective Cinchona alkaloid based organocatalyst for the
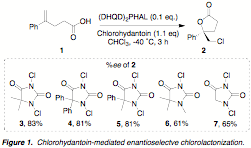
chlorolactonization of 4-arylpentenoic acids (Figure 1). In the presence of only 10 mol % of
(DHQD)2PHAL, alkenoic acid 1 is cyclized to give chloro-g-lactone 2 in high yield and 83% ee, thus establishing a quaternary
chiral center. Central to the
success of this approach was the optimization of both solvent and halogen
source. Chloroform was found to be
ideal, with other solvents providing substantially reduced selectivities. The choice of
1,3-dichloro-5,5-dimethylhydantoin (3) as the halogen source proved to be crucial to provide the
highest selectivities.
Chlorination by action of N-chlorosuccinimide (NCS) returned
chlorolactone 2
with reduced ee
(65%), while bromolactonization with NBS returned the corresponding
bromolactone with 35% ee. An exhaustive screen of
other commercially available Cinchona alkaloids and derivatives thereof confirmed (DHQD)2PHAL
as the optimal catalyst. Figure 1
illustrates the screening of other chlorohydantoins and the %ee's afford by
each for the preparation of 2. The next step
is to screen a number of peptidic scaffolds such as 8 to investigate whether or not they
are capable of inducing asymmetry in the aforementioned reaction. To date, we have synthesized over 20
peptides that will be screened under different reaction conditions for the
latter purpose.
Preliminary experiments have
uncovered a viable, selective Cinchona alkaloid based organocatalyst for the
chlorolactonization of 4-arylpentenoic acids (Figure 1). In the presence of only 10 mol % of
(DHQD)2PHAL, alkenoic acid 1 is cyclized to give chloro-g-lactone 2 in high yield and 83% ee, thus establishing a quaternary
chiral center. Central to the
success of this approach was the optimization of both solvent and halogen
source. Chloroform was found to be
ideal, with other solvents providing substantially reduced selectivities. The choice of
1,3-dichloro-5,5-dimethylhydantoin (3) as the halogen source proved to be crucial to provide the
highest selectivities.
Chlorination by action of N-chlorosuccinimide (NCS) returned
chlorolactone 2
with reduced ee
(65%), while bromolactonization with NBS returned the corresponding
bromolactone with 35% ee. An exhaustive screen of
other commercially available Cinchona alkaloids and derivatives thereof confirmed (DHQD)2PHAL
as the optimal catalyst. Figure 1
illustrates the screening of other chlorohydantoins and the %ee's afford by
each for the preparation of 2. The next step
is to screen a number of peptidic scaffolds such as 8 to investigate whether or not they
are capable of inducing asymmetry in the aforementioned reaction. To date, we have synthesized over 20
peptides that will be screened under different reaction conditions for the
latter purpose.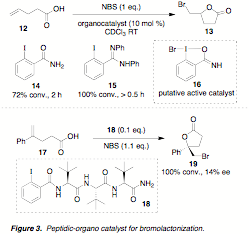 hydantoins. N-chlorohydantoins, most notably
1,3-dichloro-5,5-dimethylhydantoin (DCDMH, 3), have been used as electrophilic
chlorine sources and oxidants in a number of transfomations. We have recently disclosed a simple
methodology for the preparation of N-chlorohydantoins (10) by treatment of the hydantoin
starting materials (9) with trichloroisocyanuric acid (TCCA, 11). Ten examples were prepared in high yield, relying on a
simple recrystallization as the only purification step (Figure 2). An important benefit of this
methodology is that for the first time, chiral N-chlorohydantoins have been
prepared in high yield and enantiopurity.
These compounds are readily available by the TCCA mediated chlorination
of the corresponding chiral hydantoin substrates, which are in turn easily
prepared by known methodology from their chiral a-amino acid or amide congeners.
We were also able to demonstrate that the chlorination event and
subsequent dechlorination (by action of saturated aqueous sodium sulfite) proceeds
without erosion of enantioselectivity.
We will pursue experiments aimed at the development of
N-chlorohydantoins as reagents for asymmetric chlorination reactions.
hydantoins. N-chlorohydantoins, most notably
1,3-dichloro-5,5-dimethylhydantoin (DCDMH, 3), have been used as electrophilic
chlorine sources and oxidants in a number of transfomations. We have recently disclosed a simple
methodology for the preparation of N-chlorohydantoins (10) by treatment of the hydantoin
starting materials (9) with trichloroisocyanuric acid (TCCA, 11). Ten examples were prepared in high yield, relying on a
simple recrystallization as the only purification step (Figure 2). An important benefit of this
methodology is that for the first time, chiral N-chlorohydantoins have been
prepared in high yield and enantiopurity.
These compounds are readily available by the TCCA mediated chlorination
of the corresponding chiral hydantoin substrates, which are in turn easily
prepared by known methodology from their chiral a-amino acid or amide congeners.
We were also able to demonstrate that the chlorination event and
subsequent dechlorination (by action of saturated aqueous sodium sulfite) proceeds
without erosion of enantioselectivity.
We will pursue experiments aimed at the development of
N-chlorohydantoins as reagents for asymmetric chlorination reactions.