44181-AC10
Self-Assembly of DNA-Colloidal Systems: from Interactions to Structure
Self-assembly of structured materials from complex molecules and colloids is one of the central themes of soft condensed matter physics. Over the past decade, both the architecture of the constituent particles and the interactions between them have become much richer and more controllable, which must eventually translate into a whole new paradigm of self-assembly. In the project the conceptual and practical implications of these new possibilities are being studied, with special emphasis on micro- and nanoparticles with DNA-mediated interactions. In a typical experimental scheme, submicron colloidal particles are functionalized with certain single-stranded DNA chains (ssDNA). By changing the concentration of "linkers" (the DNA molecules whose ends are complementary to the chains attached to particles A and B, respectively) one can switch "on" and "off" the attractive force acting selectively between particles of type A and B only.
Study of random aggregation and melting. Most of the experimental studies of DNA-colloidal self-assembly to date (with rare but notorious exceptions) do not produce any structure with long-range order due to glassy dynamics. However, the observed self-assembly of disordered aggregates does carry important information about the underlying interparticle interactions. In a recent work, we employed the effective potential method to calculate the melting profile in the regime of reversible aggregation of colloids with DNA--mediated interactions. This macroscopic behavior was described starting with the known microscopic properties of the system, such as DNA coverage and hybridization free energy. In a joint study with experimentalists form NYU, we have demonstrated that the melting point can be tuned in a very controllable way, and the parameters of aggregation transition are in a good agreement with the theory.
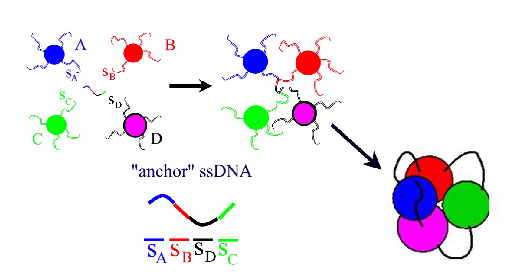
Programmable self-assembly of nanoparticle clusters and DNA-caged nanoparticles. As an alternative to the traditional bulk self-assembly, we proposed to build relatively small particle clusters of prescribed geometry. These clusters can be subsequently used in hierarchical self-assembly schemes as building bocks for larger structures. An alternative to building the nanoparticle clusters is to self-assemble "caged" particles. These are nanostructures consisting of a single particle surrounded by DNA "cage" of prescribed geometry. We have theoretically proposed the schemes and identified the optimal regimes for for building such structures.
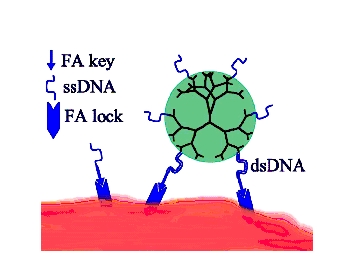
The proposed scheme of programmable self-assembly of micro/nanoclusters. What makes "hairy particles" slow? In order to facilitate the self-assembly of ordered DNA-colloidal structures one has to understand the origin of the experimentally observed slow relaxation dynamics in these systems. We studied the kinetics of particles which can form multiple "key-lock" bonds with certain substrate, or other particles [key1]-[key2]. Since the number of bonds is random and depends on the relative orientations and positions of the interacting particles, the overall dynamics is a complex interplay of lateral particle diffusion and the binding-unbinding processes. Our study reveals a number of peculiar properties of this model system. First, there is a wide power-law-like distribution of the relaxation times, consistent with experimental data. Second, the lateral particle motion is typically an anomalous diffusion. Finally, the system exhibits "aging" effect: as the particle explores the binding energy landscape by diffusion, it is able to find dipper and dipper minima and hence dramatically extend the bond lifetime. From the practical point of view, an important prediction of our theory is that the fastest dynamics is achieved when a large number of weak bonds are used instead of small number of strong ones.
Cooperative key-lock binding and drug delivery. The above model is not limited to particle with DNA-mediated interactions. Very similar ideas can be applied to other systems with key-lock binding, which may be of a great biological and medical importance. In particular, we have studied the properties of dendrimer particles decorated with functional groups capable of selective adsorption to certain membrane proteins in living cells. These nanodevices have been suggested for cell specific drug delivery, e.g. for chemotherapy. The scheme is based on the notion that certain membrane proteins (e. g. folic acid receptors) are more expressed in cancerous cells than in normal ones. This contrast should be further enhanced due to the cooperative binding: if each nanodevice can make more than one bond to target the proteins, its binding will be highly specific to cancerous cells. However, our analysis of in-vitro experiments shows that the cooperativity is kinetically limited. In other words, if the target proteins are immobilized (which is often the case in a real biological membrane), it will take many binding-unbinding events to find a configuration with sufficient number of bonds. On smaller time scales, one typically encounters particles with just one or two bonds. The non-trivial prediction of our theory is that the cooperativity and hence the specificity can be enhanced by reducing the strength of individual key-lock bonds. We have suggested a particular realization of this scenario by using the DNA-mediated scheme (see the Figure below).