

46584-G10
Inorganic Guest Species in Organic Crystals: A First-Principles Investigation
Overview and motivation
New organic electrode materials have the potential to dramatically expand the applicability of battery technology to new sectors such as automotive transport. Organic electrodes in rechargeable batteries must be able to intercalate ionic species that are either shuttled between electrodes or extracted from the electrolyte. The ability to design organic molecular building blocks with precisely defined electronic and steric features enables the synthesis of organic solids having electrochemical, kinetic and mechanical properties specially tuned for a specific battery application. However, before the rational design of such materials is possible, a fundamental understanding about the electronic, thermodynamic and kinetic properties of guest elements within various electronically conducting organic solids is essential. First-principles electronic structure methods can play a pivotal role in elucidating the mechanisms of charge transfer between electron donating or withdrawing guest species in electroactive organic crystals as well as rationalizing thermodynamic stability of and ionic mobility within organic electrode materials.
Accomplishments of the first year
During the first year of this Petroleum Research Grant, we have devoted our efforts to developing an automated statistical mechanical software tool to predict phase diagrams and diffusion coefficients in intercalation compounds with arbitrary crystallographic complexity. This tool is essential to enable a systematic study of the thermodynamic and kinetic properties of different electronically conducting organic compounds.
 As a first test case, we applied the software tool
to predict the temperature composition phase diagram and lithium diffusion
coefficient in LixTiS2. Our approach uses atomic-scale
energies from first principles to parameterize an easily evaluated cluster
expansion Hamiltonian, which can then be used in thermodynamic (Monte Carlo)
and kinetic (kinetic Monte Carlo) simulations to determine diffusion constants.
A Kubo-Green formalism is used to calculate diffusion coefficients for
interstitial diffusion in the non-dilute regime with kinetic Monte Carlo
simulations. We applied this formalism to predict the finite-temperature
thermodynamic properties of LixTiS2 as well as its
concentration dependent Li diffusion coefficients.
As a first test case, we applied the software tool
to predict the temperature composition phase diagram and lithium diffusion
coefficient in LixTiS2. Our approach uses atomic-scale
energies from first principles to parameterize an easily evaluated cluster
expansion Hamiltonian, which can then be used in thermodynamic (Monte Carlo)
and kinetic (kinetic Monte Carlo) simulations to determine diffusion constants.
A Kubo-Green formalism is used to calculate diffusion coefficients for
interstitial diffusion in the non-dilute regime with kinetic Monte Carlo
simulations. We applied this formalism to predict the finite-temperature
thermodynamic properties of LixTiS2 as well as its
concentration dependent Li diffusion coefficients.
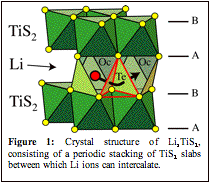
The sulfur atoms of LiTiS2 form two-dimensional triangular lattices stacked in an ABAB sequence, leading to a hexagonally close-packed anion sublattice. The Ti and Li ions alternately fill octahedral sites between the close-packed sulfur planes. The crystal can be viewed as an array of TiS2 slabs separated by layers of Li (see Fig. 1)
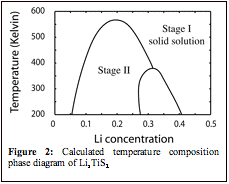
The electrochemical removal of Li from LiTiS2 results in the creation of Li-vacancies between the TiS2 slabs, introducing configurational disorder. Monte Carlo simulations applied to the cluster expansion allows us to construct a phase diagram depicting phase stability as a function of Li concentration and temperature. Figure 2 illustrates the calculated phase diagram for LixTiS2 between 200 K and 600 K..
Li diffusion in intercalation compounds occurs by a vacancy mechanism, whereby individual Li ions hop into neighboring vacant sites. Crystallographically, the least constricted pathway for Li migration between neighboring octahedral sites of layered transition metal sulfides having the O1 crystal structure is through an adjacent tetrahedral site. This is schematically illustrated in Fig. 1.
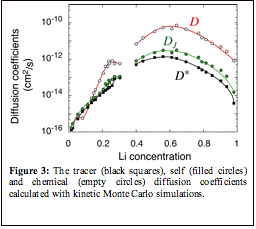 To
calculate diffusion coefficients at finite temperature, we used a standard
kinetic Monte Carlo algorithm. Figure 3 illustrates calculated diffusion
coefficients at 300 K as a function of Li concentration. As is clear from Fig.
3, the diffusion coefficients all have a very strong dependence on Li
concentration, exhibiting a maximum close to x=0.5 and varying by several
orders of magnitude over the whole concentration interval. The diffusion
coefficients, decreases rapidly as the Li concentration is reduced below
x=0.55. The kinetic Monte Carlo simulations show that Li transport is dominated
by hops into divacancies. The migration barriers associated with divacancies
are significantly lower than those into isolated vacancies (which involve
tetrahedral sites that share a face with an occupied octahedral site). However,
the divacancy concentration is very low at high Li concentration, increasing
with decreasing Li concentration. Below x=0.55, the diffusion coefficients
decrease in spite of the continued increase in the number of divacancies. This
decrease in the diffusion coefficients results from an increase in the
migration barriers at low Li concentration.
To
calculate diffusion coefficients at finite temperature, we used a standard
kinetic Monte Carlo algorithm. Figure 3 illustrates calculated diffusion
coefficients at 300 K as a function of Li concentration. As is clear from Fig.
3, the diffusion coefficients all have a very strong dependence on Li
concentration, exhibiting a maximum close to x=0.5 and varying by several
orders of magnitude over the whole concentration interval. The diffusion
coefficients, decreases rapidly as the Li concentration is reduced below
x=0.55. The kinetic Monte Carlo simulations show that Li transport is dominated
by hops into divacancies. The migration barriers associated with divacancies
are significantly lower than those into isolated vacancies (which involve
tetrahedral sites that share a face with an occupied octahedral site). However,
the divacancy concentration is very low at high Li concentration, increasing
with decreasing Li concentration. Below x=0.55, the diffusion coefficients
decrease in spite of the continued increase in the number of divacancies. This
decrease in the diffusion coefficients results from an increase in the
migration barriers at low Li concentration.
Future work
With the automated statistical mechanics software tools developed during the past year, we are in a position to study the thermodynamic and kinetic properties of Li and anion insertion in a variety of electronically conducting organic crystals. During the next year, we will investigate the equilibrium voltage curve and guest ion diffusion coefficients in three candidate organic electrode materials: polyaniline, polythiophene and polypyrrole.