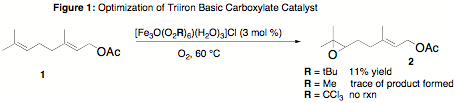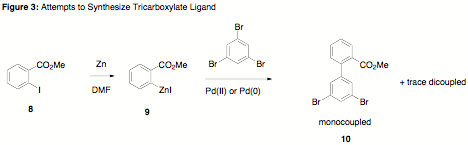46714-G1
Remote Functionalization Reactions Catalyzed by Multinuclear Complexes
Studies Conducted: A previous report described the efficient oxidation of organic substrates using TBCs that are consistent with an ester carbonyl of geranyl acetate (1) directing remote oxidation. The first round of experiments conducted on this project were geared towards reproducing the published experiments and optimizing the catalyst structure for future experiments. In these studies, three TBCs of the general formula [Fe3O(O2CR)6(H2O)3]Cl were prepared for this purpose by literature methods. These catalysts were derived from pivaloic (R = tBu), acetic (R = Me) and trichloroacetic (R = CCl3) acids. Numerous attempts were made to reproduce the oxidation chemistry of geranyl acetate that was reported (Figure 1, epoxide 2 in 71% yield),1 but poor conversion was noted will all catalysts. The cationic TBC derived from pivaloic acid was deemed the most reactive, so this catalyst was used in subsequent studies.
As
detailed in the original proposal, the regioselective epoxidation of C6-C7
olefin of farnesyl acetate (3), a derivative of geranyl acetate was expected to occur if
the cationic TBC [Fe3O(O2CR)6(H2O)3]Cl
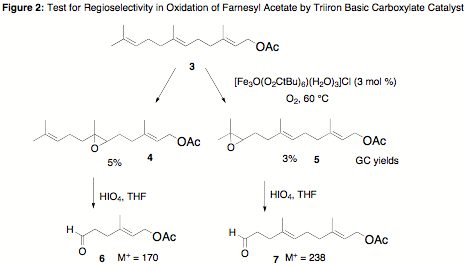
was mediating a directed oxidation reaction. Subjection of farnesyl acetate to
the same conditions furnished mostly starting material 3 with low yields of two products (4 and 5), confirming that the epoxidation
reaction was not regioselective and therefore was occurring by a directed
mechanism (Figure 2). The structures of these products, which were inseparable
by column chromatography, were assigned by degradation studies. This mixture
was subjected to oxidative cleavage by aqueous HIO4, which is known
to transform epoxides into vicinal diols that are subsequently cleaved by
periodate to aldehydes. Two products were detected by GC-MS analysis of the
crude reaction mixture, consistent with the aldehydes shown below. Due to the
low reactivity of TBCs in epoxidation reactions and the observation that the
reaction of 3
was not regioselective, this project was terminated and resources were
redirected towards a more promising project.
Design
of a new ligand for constructing trimetallic basic carboxylates was initiated
in order to test the hypothesis that stabilizing the trimetallic core, by
coordinating the metal atoms to a single ligand, one on each side of the
trimetallic core, would enhance the stability of these complexes and would
furnish better catalysts. A 1,3,5-tri(2-benzoyl)benzene scaffold was envisioned
for this purpose, and synthetic studies were initiated. The
first envisioned route to the desired ligand involved a Negishi coupling
between 9 and
1,3,5-tribromobenzene (Figure 3). Formation of the arylzinc species was
accomplished in by sonification of the aryl iodide in warm DMF in the presence
of Zn dust. Complete conversion was confirmed by quenching with HCl and
analysis by GC-MS. Subsequent Negishi coupling of the aryl zinc species with
1,3,5-tribromobenzene proved difficult, and mostly monocoupling was observed.
The
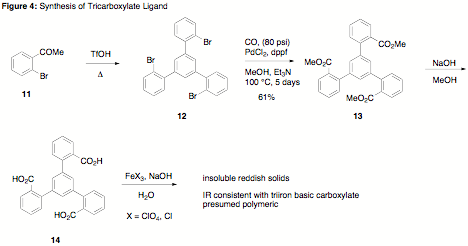
second approach to the desired ligand was successful. Trimerization of
bromoacetophenone in the presence of triflic acid produced the tetraaryl
tribromide 12 as
reported in the literature.2 Subsequent esterfication of the bromide
by palladium-catalyzed carbonylation provided the target triester 13. A reasonable yield of 13 was obtained after higher
temperatures (100 °C) and longer reactions times (5 days) were employed.
Attempts to optimize this reaction further by incorporating other Pd catalysts
were unsuccessful. According to TLC analysis, the first and second
carbonylation were fast, whereas the third was slow, presumably due to the
crowded steric environment of the ligand. Saponification of triester 13 was straightforward, and the
neutral triacid was obtained as a crystalline colorless solid. Synthesis of two
triiron basic carboxylates was carried out using FeCl3 or Fe(ClO4)3
as the iron source. Insoluble materials, consistent with the formation of
polymeric iron carboxylates, were observed. The IR spectra of the solids were
consistent with formation of triiron basic carboxylates, as evidenced by a
change in the C=O stretch. The insolubility of these complexes prevented their
testing as homogeneous catalysts.
References: ADDIN EN.REFLIST (1) Ito, S.;
Inoue, K.; Mastumoto, M. [Fe3O(OCOR)6L3]+-catalyzed epoxidation of olefinic
alcohol acetates by molecular oxygen. Journal of the American Chemical
Society 1982, 104, 6450-2. (2) Feng, X.;
Wu, J.; Enkelmann, V.; Muellen, K. Hexa-peri-hexabenzocoronenes by efficient
oxidative cyclodehydrogenation: The role of the oligophenylene precursors. Organic
Letters 2006, 8, 1145-1148.