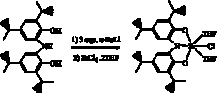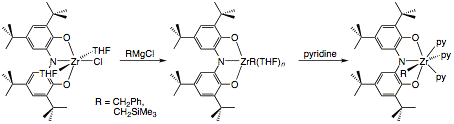45941-AC3
Harnessing Metal-Ligand Cooperative Redox Reactivity for Bond Functionalization Strategies
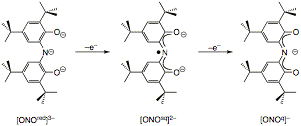
The primary focus of this project is to introduce redox reactivity to electrophilic early transition metal complexes through the use of redox-active ligands. Our previous work has focused on bidentate ortho-amidophenolate and ortho-phenylenediamide ligands coordinated to Group IV metals. In short, our resulted showed that the catechol/semi-quinone/quinone properties of these ligand platforms did in fact enable redox-based bond-activation reactions like oxidative addition and reductive elimination to occur at d0 metal centers; however, complications always arose due to dissociation of the redox-active ligands from the metal coordination sphere. To overcome this ligand dissociation problem, in the work reported here we have turned to a tridentate amido bis(phenolate) ligand, [ONOred]H3, prepared according to Scheme 1. The three oxidation states of the [ONO] ligand are shown in Scheme 2.
Scheme 1.
Scheme 2. Scheme 3. Scheme 4.
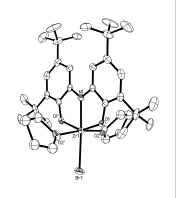
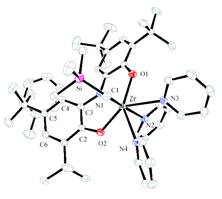
Scheme 5. Figure 1. Molecular structure of [ONOred]ZrBr(THF)2
(3).
The
zirconium monohalide complexes are suitable precursors for the preparation of
zirconium alkyl species. As shown in Scheme 6, the reaction of [ONOred]ZrCl(THF)2
with Grignard reagents RMgCl (R = CH2Si(CH3)3,
CH2Ph) afforded the expected alkyl derivatives. While the
preparation of the THF-solvated alkylzirconium complexes was straight-forward,
the purification of these species was hampered by high solubilities. Conversion
to the pyridine adducts allowed for purification and characterization by
crystallography. The surprising result, shown in Figure 2, is that [ONOred]Zr(CH2SiMe3)(py)3
(4) adopts a seven-coordinate geometry
thanks to the coordination of three pyridine molecules.
Scheme 6. Figure 2. Molecular structure of [ONOred]Zr(CH2SiMe3)(py)3
(4).
With
the successful preparation of monohalide and monoalkyl zirconium complexes of
the [ONOred]3– ligand, we have begun examining
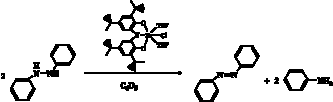
small-molecule redox reactivity. The monochloride complex, [ONOred]ZrCl(THF)2
catalyzes the disproportionation of diphenylhydrazine into aniline and
azobenzene (Scheme 7). Control reactions with Lewis acid complexes do not lead
to disproportionation indicating the importance of the redox-active ligand in
this reaction. Current efforts are aimed at elucidating the mechanism of this
reaction, which we believe proceeds through a zirconium imido intermediate,
[ONOq]ZrCl(=NPh). Such an intermediate is only possible if the [ONO]
ligand is acting as a two-electron reservoir during the reaction. Further
efforts are aimed at diverting this putative imido intermediate for catalytic
nitrene transfer reactions.
Scheme 7. 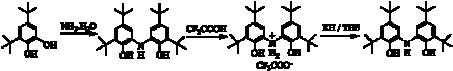

 We
have successfully metallated the [ONOred]H3 ligand
platform with zirconium and have prepared both zirconium alkyl and zirconium
halide complexes that will be used in future reactivity studies. The most
straight-forward approach towards the synthesis of a low-coordinate zirconium
complex was to treat [ONOred]H3 with tetra(alkyl)
zirconium(IV) synthons. According to Scheme 3, the dimeric complex, {[ONOred]Zr(CH2SiMe3)]2
(1), was prepared in modest yields in
this manner. The structure of 1
was confirmed by NMR spectroscopy and X-ray crystallography. The surprising
feature of the structure is the bridging coordination mode for the chelating
[ONOred]3– ligands. The Zr–N bond distances
are long at 2.31 and 2.50 ü, consistent with each amide acting as a donor to
two different zirconium centers. The preparation and characterization of 1 as a dimer was an important result because it
highlights the difficulty associated with targeting low-coordinate zirconium
complexes with the [ONOred]3– ligand. As such we
began to target complexes that were five- or six-coordinate, but with weakly
coordinating solvent molecules that could be removed under mild conditions.
We
have successfully metallated the [ONOred]H3 ligand
platform with zirconium and have prepared both zirconium alkyl and zirconium
halide complexes that will be used in future reactivity studies. The most
straight-forward approach towards the synthesis of a low-coordinate zirconium
complex was to treat [ONOred]H3 with tetra(alkyl)
zirconium(IV) synthons. According to Scheme 3, the dimeric complex, {[ONOred]Zr(CH2SiMe3)]2
(1), was prepared in modest yields in
this manner. The structure of 1
was confirmed by NMR spectroscopy and X-ray crystallography. The surprising
feature of the structure is the bridging coordination mode for the chelating
[ONOred]3– ligands. The Zr–N bond distances
are long at 2.31 and 2.50 ü, consistent with each amide acting as a donor to
two different zirconium centers. The preparation and characterization of 1 as a dimer was an important result because it
highlights the difficulty associated with targeting low-coordinate zirconium
complexes with the [ONOred]3– ligand. As such we
began to target complexes that were five- or six-coordinate, but with weakly
coordinating solvent molecules that could be removed under mild conditions.
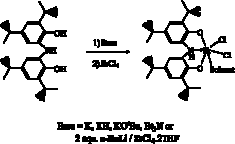
 Both
mono- and dihalide complexes of zirconium could be prepared through simple
methesis reactions. According to Scheme 4, various bases may be used to prepare
the dichloride, [ON(H)O]ZrCl2(solv) (solv = Et2O, THF,
py) (2). Complex 2 is a stable intermediate which can be isolated and
stored for long periods of time. The stability of this species derives from the
protonation state of the redox-active ligand. The amide nitrogen remains
protonated, in effect killing the redox-activity of the ligand; however, it the
redox-activity is readily unmasked by addition of another equivalent of strong
base such as nBuLi. Alternatively, we have recently been able to prepare
directly mono-halide complexes of zirconium by the direct deprotonation of [ONOred]H3
with three equivalents of nBuLi and reaction with the appropriate zirconium
halide salt in the presence of THF (Scheme 5). Figure 1 shows a the results of
an X-ray diffraction experiment on [ONOred]ZrBr(THF)2 (3). As expected, the [ONOred]3–
ligand of 3 occupies three
meridional coordination sites on the octahedral complex with the bromide ligand
trans to the amide donor. Overall the complex has approximate C2v
symmetry.
Both
mono- and dihalide complexes of zirconium could be prepared through simple
methesis reactions. According to Scheme 4, various bases may be used to prepare
the dichloride, [ON(H)O]ZrCl2(solv) (solv = Et2O, THF,
py) (2). Complex 2 is a stable intermediate which can be isolated and
stored for long periods of time. The stability of this species derives from the
protonation state of the redox-active ligand. The amide nitrogen remains
protonated, in effect killing the redox-activity of the ligand; however, it the
redox-activity is readily unmasked by addition of another equivalent of strong
base such as nBuLi. Alternatively, we have recently been able to prepare
directly mono-halide complexes of zirconium by the direct deprotonation of [ONOred]H3
with three equivalents of nBuLi and reaction with the appropriate zirconium
halide salt in the presence of THF (Scheme 5). Figure 1 shows a the results of
an X-ray diffraction experiment on [ONOred]ZrBr(THF)2 (3). As expected, the [ONOred]3–
ligand of 3 occupies three
meridional coordination sites on the octahedral complex with the bromide ligand
trans to the amide donor. Overall the complex has approximate C2v
symmetry.