47159-GB4
The Scope and Chemical Relevance of Anion-Pi Interactions Involving Aromatics: Computational and Solid-State Studies
Our proposal outlined three related areas of research on the interaction between anions and aromatic π-electron density. We have made significant progress on all three fronts, as summarized below.
Correlation Between Anion-Arene Binding Energies and the Polarizability of the Aromatics. We have calculated the anion-arene binding energies between halo-substituted aromatics and Cl– and Br– anions and found an excellent correlation with the sum of the Hammett substituent constants σp (Σ σp) of the substituted aromatics. This is an exciting result, and it coincides with recent studies showing correlation between Hammett substituent constants and either anion-arene[1] or arene-arene[2] binding energies. Given that dispersion and polarizability are both known to be factors that contribute to the binding between anions and aromatic π-electron density, this result suggests that Hammett σp values describe the polarizability of substituted aromatics. Our ongoing efforts in this area involve calculating the polarizability values (<α>) of the substituted aromatics, determining how broad the correlation is between anion-arene binding energies and the Σ σp values of substituted aromatics, determining if a correlation exists between anion-arene binding energies and the < α> values of substituted aromatics, and determining why σp values seem to be indicative of aromatic polarizability.
Anion Binding to Transition Metal-Complexed Aromatics. We have calculated the Cl– and Br– binding of Ar-FeII-Cp complexes (Ar = substituted aromatic; Cp = cyclopentadienyl anion) where the anion binds to the π-face of the substituted aromatic Ar and compared that to the anion binding to the FeII center of the complex. We determined the preference for anion-π binding by subtracting the binding energy when the anion is complexed to the metal from the binding energy when the anion is complexed to the aromatic π-cloud. This gave the very surprising result that the preference for anion-π binding increased with an increase in electron donating groups on the aromatic. A preliminary explanation for this involves the charge on the FeII center. The greater the electron donating ability of the substituent, the greater the charge transfer to the FeII, thus increasing the anion-π binding energy and decreasing the anion-metal binding. Our work in this area has also shown a great correlation between the anion-π binding energy and the Σ σp values of the substituted Ar group. Ongoing studies in this area include explaining why electron-donating groups enhance anion-π binding in Ar-FeII-Cp complexes, expanding the studies to include Ar-RuII-Cp, Ar-FeII(CO)3 and Ar-RuII(CO)3 complexes, and including more polarizable anions.
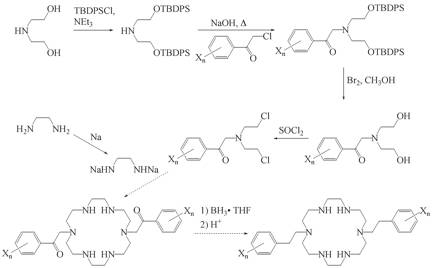
Preparation of Solid-State Host for Anion--π Binding Studies. The figure below shows the synthetic approach being employed to prepare a solid-state host to study anion-π binding. All but two of the steps have been successfully completed and the products of the completed steps have been characterized by NMR and mass spectroscopy. We are currently repeating, and improving the yields, of all reactions leading up to the marcocyclization reaction because this is expected to be a low-yield reaction. We expect to have the desired parent product, where the aromatics have all H atoms, prepared shortly. The synthetic approach will allow for facile preparation of macrocycles with differently substituted aromatic appendages via the wide availability of arene-substituted chloro- and bromo-acetophenones, which are introduced in the second step. Once we have numerous substituted analogs prepared we will begin crystallographic studies to determine what aromatic substitution patterns allow for anion-π binding in the solid-state.