

46492-GB1
Catalytic Asymmetric Methods for the Conversion of Petroleum Products Derived from Ethylene and Propylene into Chiral N-Heterocycles: the Preparation of Chiral Pyrrolidines and 1,2,3,4-Tetrahydroquinolines
Investigation of Catalytic Methods for the Preparation of N-Heterocycles.
Figure 1. Examples of bioactive quinolines.
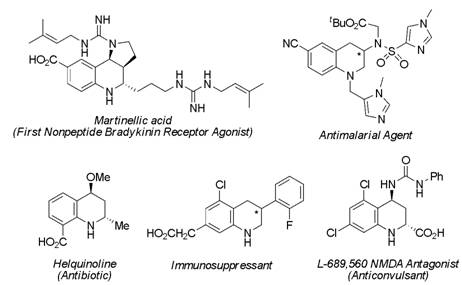
A Tandem Michael-Aldol Reaction for the Preparation of Dihydro-quinolines. 1,2,3,4-Tetrahydroquinolines and dihydroquinolines are common structural motifs in many biologically-active molecules, many of which have industrial and medicinal applications (Figure 1). Due to the importance of this compound class, the development of synthetic routes for the preparation of these compounds is of great synthetic interest.
Our initial route to access these compounds was to employ a,b-unsaturated aldehydes with proline-based iminium ion catalysis in a tandem Michael-aldol reaction. Unfortunately, a recent report by Wang and coworkers, in which the same catalyst and substrate scope were utilized, was published during our investigations of this route. As a result, we focused our subsequent efforts on the study of some unusual reactivity that we had observed during our related studies of the metal-catalyzed version of this reaction. Through considerable metal screening, we determined that copper(II) triflate (10 mol%) could catalyze the tandem Michael-aldol reaction of cinnamaldehyde 9a with a 2-N-Cbz-protected benzaldehyde 8, albeit in low conversion (19% after 48 h; Scheme 1). No conversion was observed when triflic acid, a compound commonly present in metal triflate reactions, was employed as the reaction catalyst, indicating that catalysis is metal-based. The use of a strongly-donating solvent, such as acetonitrile, proved crucial to reactivity; no conversion was observed in the presence of a noncoordinating solvent. Optimization of the Michael acceptor led us to investigate a,b-unsaturated N-acyl pyrrole 9b, which afforded the desired Michael-aldol product in 43% conversion after 48 h. Unexpectedly, the corresponding phenyl ketone—which has previously been used interchangeably with N-acyl pyrroles in organometallic reactions—afforded no reaction. The best results with this catalyst loading (93% conversion after 48 h) were obtained when the reaction was conducted at reflux. Interestingly, negligible reactivity was observed with other metal triflate salts commonly used in Michael additions (Yb(OTf)3, Sc(OTf)3, In(OTf)3, and Zn(OTf)2).
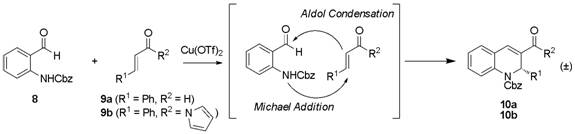
Scheme 1. Tandem Michael-Aldol Reaction.
Another feature of this reaction is that the addition of desiccants (e.g. 4 Ĺ powdered sieves, etc.) shut down reactivity, indicating that the aqueous byproduct of this reaction is tolerated. The use of other additives (bases, etc.) also proved detrimental to reactivity. To date, these reactions have proven to be remarkably clean, with GC conversion data correlating with isolated yield. Reaction scope has been extended to include several aromatic and aliphatic substrates, the products of which have been characterized via NMR and high-resolution mass spectrometry.To our knowledge, the unusual selectivity of this reaction for N-acyl pyrroles in the presence of carbamate-protected nucleophiles is unprecedented, and stands in marked contrast to previous reports. Product characterization and computational studies into the selectivity of this reaction are currently underway. We seek to submit this work for publication in the Tetrahedron by December 2008.
It is important to note that research on this project has been conducted entirely by undergraduates: lead discovery of the copper triflate system was performed by Claire Knezevic (Scripps '08); reaction optimization is currently being conducted by Anna Wagner (CMC '09) as part of her Keck-funded summer research and future senior thesis. Nicole Duggan (Scripps ‘10) is also assisting. Following the completion of the initial phase of this project, further studies into the development of an enantioselective variant of this reaction will be investigated. Given the proven pharmacology of this compound class, an undergraduate project to investigate the bioactivity of these molecules may also prove worthwhile.
1,3-Dipolar
Cycloadditions of Aziridines with Alkenes for Enantio-selective Pyrrolidine
Formation. Aziridines
are versatile building blocks for the preparation of nitrogen-containing
compounds. Their ready availability, coupled with their propensity to undergo
regio- and stereoselective ring-opening, have made them attractive targets for
organic synthesis. Numerous procedures have been developed for the ring-opening
of aziridines with various nucleophiles; however, examples of aziridine
ring-opening utilizing alkenes as nucleophiles are relatively rare (Scheme 2).
Early examples of this reaction class required heat and strained alkene ring
systems to promote reaction. More recent work has demonstrated that, by
utilizing dual-activated aziridines in the presence of a Lewis acid, efficient
conversion can occur at reduced reaction temperatures. To date, we have
investigated several scandium and ytterbium-based chiral catalysts in this
reaction. Conversion to the desired products was observed. Unfortunately,
chiral separation proved unsuccessful using our Chiralcel OD and Whelk-O HPLC
columns. With the acquisition of three new chiral HPLC columns this summer, we
hope to reinvestigate this project in the 2008-2009 academic year. Research
into the lead discovery phase of this project was conducted by Casey Smirniotopoulos
(CMC '08) as a
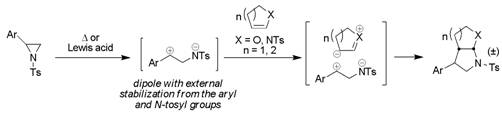
Scheme 2. 1,3-Dipolar cycloadditions of aziridines with heteroatom-substituted alkenes.