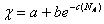46082-AC7
Basic Mechanisms Responsible for Morphology Control within Polymer Nanoparticles
Understanding morphology control for waterborne, multiphase, polymer nanoparticles is an ongoing endeavor. Upon consideration of composite nanoparticles produced by emulsion polymerization, many scholars currently accept an anchoring mechanism in which initiator end groups remain at a particle's surface. Of course this means that to fill in the composite particle, polymer chains must “tether” from the particle surface.
Figure 1: Schematic of
2nd stage polymer chain tethering within a composite latex particle. Considering the free energy required to
stretch a polymer chain through a particle, it seems more reasonable to
conclude that radical end groups either diffuse within a particle (unanchored)
or become buried by new forming polymer.
It is also important to be able to understand the solubility
characteristics of an oligoradical with a polar initiator end group within a
second polymer phase. Its solubility
within both the aqueous and polymer phases is critical for investigation of
oligoradical diffusion within a particle.
Upon
pursuing a thermodynamic investigation for testing the validity of chain
tethering, it is clear there are three factors to consider: chain stretching distance, energy required
for combining ions (thus enabling an ionic initiator end group to enter the
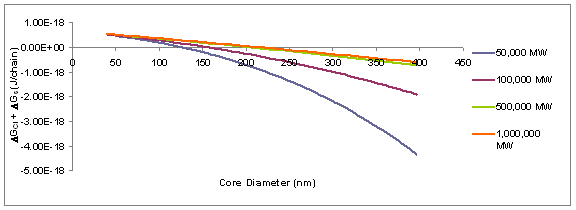
particle), and mixing enthalpy between the two polymer phases. Calculations, seen in Figure 2, have shown
that combining ions is favorable for larger particles and shorter molecular
weights.
Figure 2: Free energy
calculations for a composite latex particle.
The energy axis is the overall free energy (stretching, combining ions,
and mixing). If growing oligomeric chains are
to release from the surface of the particles and diffuse inwards, they must be
soluble in the host matrix phase (seed particle). At very short lengths these chains have a
hydrophilic end group (e.g. -SO4)
and are insoluble. As more units are
added, the chain becomes more hydrophobic and quickly changes its character,
perhaps becoming soluble in the host matrix.
As the chain length is further increased, phase separation may occur
when the growing polymer chain and host polymer are incompatible at high molecular
weights. We have attempted to describe
such solubility phenomena by calculating free energy changes upon mixing of the
growing oligomer chains and the host polymer.
Here we have taken the enthalpy term to be χ φ1φ2
as usual but write χ as
where a, b and c are constants
which depend upon the experimental system and NA is the number of
repeat units in the growing oligomer.
After extensive parameter analysis, it seems that “a” represents the
interaction parameter between both polymers at infinite chain length. The “b” constant seems to represent the
contribution of the chain end group associated with the initiator. The “c” constant is related to how quickly new
repeat units change the polarity of the growing oligomer. This has the effect of starting with χ
= a + b at NA = 0 (only the initiator end group – here we
take b >> a) and finishing with χ = a at NA = large. For realistic polymer systems (e.g. PS/PMMA)
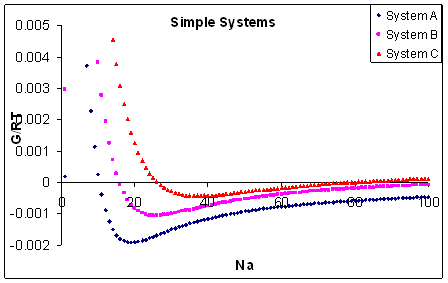
this creates ΔG vs NA curves that
appear as in Figure 3. The free energy
change is positive at small chain lengths, negative at intermediate chain
lengths and again positive at much higher chain lengths. This suggests that the growing chain is insoluble
in the host matrix until it propagates enough to become non-polar enough to be
compatible with the host polymer. It can
then diffuse (and grow) within the host matrix until its increasing chain
length creates such a loss in entropy that it phase
separates. Figure 4 shows solubility
results for oligomers of styrene with a HO3S-MNA structure
as they are mixed with methanol. These
are the first data we are aware of that show the type of solubility changes
with chain length that we computed as described above.
Figure 3: Free energy of
mixing oligomers of polymer A in a host, high MW, polymer B as the chain length of polymer A increases. SHAPE
\* MERGEFORMAT 3.0 n = 3.0 n =
3.0 n = 4.5 n = 5.5 n = 10.0 n = 15.4
Figure 4 : Styrene oligomers (HO3S-Mn)
of different chain lengths, n, mixed with methanol.
The most important results are to
come from electron spectroscopic imaging (ESI) TEM images of composite latex
particles from which an elemental map across the particle of initiator end
groups (sulfur) can be obtained. These
experiments are designed such that the surfactant does not contain sulfur, allowing
for a detailed image of the initiator distribution in the particles. In addition we are doing phase separation
experiments for the HSO3 - MNA oligomers in high molecular weight polymers, including
polystyrene and (separately) poly (methyl methacrylate) to determine the
correct nature of the Flory-Huggins interaction parameter for the growing
oligomer within the host polymer particle.