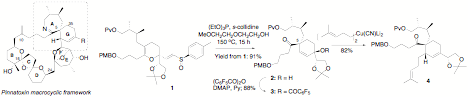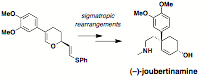43838-G1
The Tandem Claisen-Mislow-Evans Rearrangement
During the overall funding period several projects have been initiated and important accomplishments have been achieved. These results have been essential for starting my independent career, engaging graduate students in research, and securing independent major funding from other agencies. With the help of this grant, graduate students have been able to join the project and spend more time in the laboratory. As evidence of effectiveness of the grant in this regard, seven publications with graduate students (including first-year graduate students) and postdoctoral fellows have been published, and two more future publications (in preparation) will acknowledge the support of the ACS PRF grant.
The research performed with the support of the PRF grant is an interplay of the development of new synthetic methodology and the total synthesis of natural products. We developed two new variations of the classic sigmatropic rearrangements as a potential solution to the challenge posed by the structure of marine alkaloid pinnatoxin A. The first reaction is a tandem Claisen-Mislow-Evans rearrangement that efficiently formed the quaternary chiral center at the core of the characteristic spiroimine of pinnatoxin A (Scheme 1). However, we were unable to advance the product further toward pinnatoxin A due to undesired stereochemistry in the cuprate substitution of the allylic pentafluoroacetate 3. The tandem Claisen-Mislow-Evans rearrangement will be employed in the synthesis of other natural products containing a cyclohexenol moiety.
Scheme
1. This outcome prompted a
change of strategy that stimulated the development of the diastereoselective
Ireland-Claisen rearrangement of alpha-branched esters through stereoselective
enolization. This type of enolization could not be achieved before in acyclic
systems by known methods. The stereodefined, tetrasubstituted enolates are
potentially useful for a variety of asymmetric transformations. We initially
focused our efforts on the Ireland-Claisen rearrangement and its application in
the total synthesis of pinnatoxin A and related natural products. We found that the
stereoselectivity of enolization of acyclic, alpha-branched chiral esters can
be efficiently controlled using chiral lithium amide bases. We employed widely
available bis-(1-phenylethyl)amine and Koga amines as the precursor to the
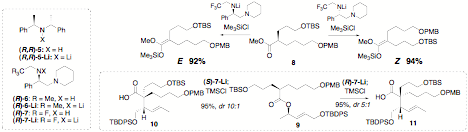
chiral lithium amides. We found that amines 5-7 are
generally effective and afford good to excellent selectivities with a range of
substrates. For example, enolization of ester 8 with amide 7-Li is highly stereoselective, and the configuration of the enolate is
controlled by the chirality of the base. Application for the Ireland-Claisen
rearrangement of allylic ester 9
gives ester 10 or 11 stereoselectively, with the sense of selectivity
once again controlled by the chirality of the base. Using standard methods (LDA
etc.), an equimolar mixture of 10
and 11 is produced.
Scheme
2. In short, this methodology
allows for a rapid build-up of complexity from readily available chiral
carboxylic acids and chiral allylic alcohols via esterification and
Ireland-Claisen rearrangement forming congested chiral centers which would be
difficult to access by known methods. This methodology is
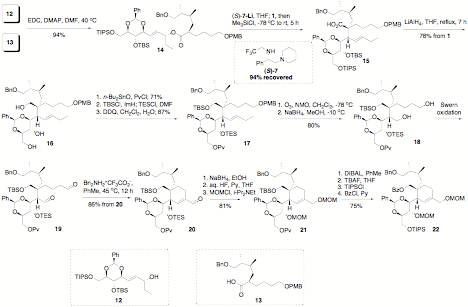
highlighted in our synthesis of the spiroimine fragment of pinnatoxins. As
shown in Scheme 3, condensation of alcohol 12 and acid 13, prepared easily by standard methods, followed by the Ireland-Claisen
rearrangement afforded 15 in high
yield and diastereoselectivity, validating our approach. The reaction performed
very well giving identical yield when carried out twice on 2.1 g scale. The
product was advanced efficiently to dialdehyde 19, which was cyclized by a high-yielding aldol
condensation to form the cyclohexene ring of pinnatoxins. Subsequent
manipulations revealed that a better protecting group strategy is desireable.
Studies toward this goal are currently underway in our laboratory. We have also prepared the
BCD-dispiroketal fragment of pinnatoxins, using the opportunity to study the
solvent effect on the thermodynamically controlled selectivity of the
acid-catalyzed spiroketalization reaction. This study provided sufficient
quantity of this building block to advance further towards the target molecule,
which will be a central goal for the next year. The tandem Claisen-Mislow-Evans
reaction developed earlier served as an foundation for the enantioselective
total synthesis of two Sceletium
plant alkaloids (–)-joubertinamine and (–)-mesembrine (Scheme 4).
This effort resulted in the first enantioselective synthesis of joubertinamine,
which will be published in the special issue of Tetrahedron in recognition of Professor Du Bois' contributions
to organic synthesis. In conclusion, we the
critical support of the ACS PRF fund, we were able to establish a research
program in the area of synthetic organic chemistry that combines the
development of new synthetic methods and their application in the synthesis of
complex natural products. The importance of this support is underscored by the
fact that this is a sole source of external funding at the beginning of our
program.
Scheme
3.
Scheme
4.