

44534-B3
Sites of Protonation and Ligand Migration in Bimetallic Organometallic Complexes
This report focuses on a computational chemistry study of some bimetallic organometallic complexes. Our computational modeling is closely connected to the experimental work performed in the laboratories of Dr. M. Cowie at the University of Alberta, Edmonton.
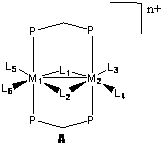
The purpose of this work is to examine fundamental catalytic conversions that occur at the bimetallic centers. The general structure is shown below in A, where the metal centers are from groups 8 and 9 in the periodic table. Though not involved in the chemistry in our study, the diphenyldiphosphinomethane ligand (dppm) provides the structural support necessary to hold the two metals in place. Around the metals are ligands occupying positions L1-L6, involved more directly in this study. Not all of these positions are necessarily occupied at all times, with positions varying based on the system being studied. The ligands involved include acetyl, carbonyl, hydride, methyl, methylene, phosphine, and triflate anion. The overall charge of the complexes includes neutral, cationic, and dicationic, depending on the ligands and metals involved.
Gaussian03 was used for all calculations. The B3LYP theoretical method was employed for all calculations. The cc-pVDZ basis set was utilized for all atoms except the transition metal atoms, for which the LANL2DZ basis set was employed. The dihydrogen diphosphine methane ligand (dhpm) was utilized in place of the dppm ligand for our modeling work.
We have examined seven sites of protonation and a hydrogen atom migration mechanism on [RhOs(CO)3PH3(μ-CH2)(dhpm)2]+. Hence, the protonated molecule is a dication. The seven structures are depicted here.

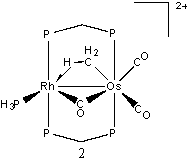
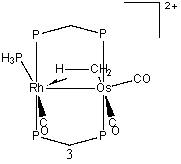



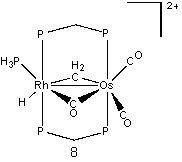
Structure 1 shows the bimetallic complex in its unprotonated form. Each protonation occurs on this structure. The following seven structures are optimized structures of 1 after protonation. The most stable protonated structure is 2, which is a µ-CO structure with methylene group, and corresponds to the protonation of the bridging methylene (and corresponds to a structure observed by Cowie, et al.) The energies of the remainder of the structures will be reported relative to this structure. There was another result when the hydrogen atom was added to the bridging methylene. In this scenario, no µ-CO was formed, and the structure obtained (3) had an energy of 5.3 kcal/mol. The second site of protonation was on the rhodium atom, 4. This resulted in the bridging carbonyl moving into a semi-bridging position, with an energy of 12.3 kcal/mol. Next, protonation was attempted on the osmium atom, 5, behind both carbonyl groups. This structure had a relative energy of 16.3 kcal/mol.
The next site of protonation was in the open area between the metals, opposite the methylene ligand. Protonation here led to the formation of a hydride bridge, 6.
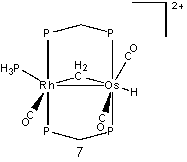
We also attempted protonation between the carbonyls on the osmium, 7, with a relative energy of 18.5 kcal/mol. Similar to the protonation on the osmium, 5, a carbonyl group moved into a semi-bridging position opposite the µ-CH2. The high relative energy for this was somewhat surprising based upon the HOMO, which shows high electron density around the two carbonyl groups. Finally, protonation was attempted on the rhodium atom between the carbonyl and phosphine groups. The resulting structure, 8, had the two ligands nearly 180˚ apart from each other. The carbonyl moved into a bridging position as well. Despite this, the structure had a relative energy of 27.0 kcal/mol.
The protonation sites on the bridging methyl ligand and the hydride bridge were the most stable. 2 has been found experimentally by Dr. Cowie; it is perceived that the other two (3 and 6) are then intermediates by which 2 is formed.
T1 is a transition state between structures 2 and 3. Three movements occur: the carbonyl on rhodium moves into a bridging position between the metals, the phosphine ligand swings from behind the Rh-Os-dhpm plane to a position in front of the Rh-Os-dhpm plane, and the methyl moved from directly behind the osmium to a bridging position between the two metals.

The transition state from 6 to 2 has been more difficult to find. Using QST3, a transition state was thought to be found (T2). This had the hydrogen positioned directly between the metals, with the metals moved apart slightly. The relative energy of T2 was found to be 40.8 kcal/mol. A frequency was run on this transition state, and three imaginary frequencies were found. One of these was found to be negligible, as it involved the twisting of the phosphine ligand. The other two involved the hydrogen moving up and down between the metals, and in and out of the Rh-Os-dhpm plane. Therefore, it is a second order saddle point and not a true transition state. Further studies are underway to learn more about this transition state.