

45988-AC6
Understanding Chemical Bond Dynamics in Solution using Femtosecond Spectroscopy and Nonadiabatic Mixed Quantum/Classical Simulations
On the theoretical side of the project, graduate student Will Glover has
developed a new method for performing mixed quantum/classical
simulations using CISD to accurately describe the quantum behavior of
multiple electrons in molecular systems. Our approach has been to
express the electronic wavefunction directly on a six-dimensional
real-space grid, of size 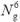 , with Ng = 24 or 32. We thus solve
, with Ng = 24 or 32. We thus solve 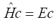 where c is the vector of expansion coefficients of the electronic
wavefunction in the six-dimensional basis and E is the energy eigenvalue. The full matrix representation of H would require the storage of
where c is the vector of expansion coefficients of the electronic
wavefunction in the six-dimensional basis and E is the energy eigenvalue. The full matrix representation of H would require the storage of 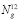 elements, which rapidly becomes impractical. Instead, we use an
iterative subspace algorithm that only requires the operation of H
on a six-dimensional vector. This allows us to store the potential energy matrix elements in real-space and the kinetic energy matrix
elements in momentum-space, reducing the storage requirements to
two diagonal matrices, which scales only as
elements, which rapidly becomes impractical. Instead, we use an
iterative subspace algorithm that only requires the operation of H
on a six-dimensional vector. This allows us to store the potential energy matrix elements in real-space and the kinetic energy matrix
elements in momentum-space, reducing the storage requirements to
two diagonal matrices, which scales only as 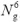 . Transformations between real-space and momentum-space are achieved with six-dimensional fast
fourier transforms (FFTs). This provides the additional advantage that the
computational effort of the FFT scales as
. Transformations between real-space and momentum-space are achieved with six-dimensional fast
fourier transforms (FFTs). This provides the additional advantage that the
computational effort of the FFT scales as 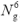 Log Ng, such that the cost of
operating the Hamiltonian on a 6D vector scales almost linearly with the
number of grid points.
Log Ng, such that the cost of
operating the Hamiltonian on a 6D vector scales almost linearly with the
number of grid points.
 Our first application of the new method has been to describe the dynamics of Na2 in classical liquid Ar. The bonding electrons are treated fully
quantum mechanically with our CISD algorithm (which is exact for two
electrons), and the other interactions are accounted for using
pseudopotentials. We find a significant shift of the bond vibration
frequency of the dimer between the gas phase and solution. Although
such a shift is expected, there is no way to account for it by simply grafting the gas-phase potential energy surface (PES) into a solution-phase
simulation: the shape of the PES is altered by the fact that the solvent pushes on the bonding electrons, changing the interaction between the
Na nuclei. Another consequence of the interaction between the solvent
and the bonding electrons is the fact that the normally non-polar Na dimer
develops a significant dipole moment in solution (~3 Debye), even in a
perfectly non-polar solvent such a liquid Ar. This is due to short-range repulsive forces from the solvent that are constantly "squishing" the
bonding electrons, so that the electron cloud is not centered on the nuclei. This type of "non-polar polarizability" has not been previously reported,
and should have enormous consequences for the behavior of such
systems in response to static or oscillating electric fields, including
photons. We also find that the quantum description of the bonding
electrons leads to a solvent structure that cannot be reproduced
classically. The left side of the TOC graphic shows the pair distribution
function for solvent molecules as distributed around the dimer center of mass; the z-axis of these cylindrically-averaged functions is the
internuclear bond axis, and the r-axis points along the bond bisector.
The right side of the TOC graphic shows the results of a classical
simulation where Na2 is described as two Lennard-Jones spheres
with the appropriate diameter and bond length (i.e., a `dumbbell' molecule).
The classical simulation gives an incorrect structure with
solvent preferentially lying in the small gap between the two atoms, where
the quantum simulation shows solvent is unlikely to go. Thus, a correct
description of solvent structure and dynamics requires that bonding
electrons be treated quantum mechanically -- classical approximations,
even highly sophisticated ones, simply cannot do the job, because they
cannot correctly account for the interaction of the solvent weith the bonding electrons
Our first application of the new method has been to describe the dynamics of Na2 in classical liquid Ar. The bonding electrons are treated fully
quantum mechanically with our CISD algorithm (which is exact for two
electrons), and the other interactions are accounted for using
pseudopotentials. We find a significant shift of the bond vibration
frequency of the dimer between the gas phase and solution. Although
such a shift is expected, there is no way to account for it by simply grafting the gas-phase potential energy surface (PES) into a solution-phase
simulation: the shape of the PES is altered by the fact that the solvent pushes on the bonding electrons, changing the interaction between the
Na nuclei. Another consequence of the interaction between the solvent
and the bonding electrons is the fact that the normally non-polar Na dimer
develops a significant dipole moment in solution (~3 Debye), even in a
perfectly non-polar solvent such a liquid Ar. This is due to short-range repulsive forces from the solvent that are constantly "squishing" the
bonding electrons, so that the electron cloud is not centered on the nuclei. This type of "non-polar polarizability" has not been previously reported,
and should have enormous consequences for the behavior of such
systems in response to static or oscillating electric fields, including
photons. We also find that the quantum description of the bonding
electrons leads to a solvent structure that cannot be reproduced
classically. The left side of the TOC graphic shows the pair distribution
function for solvent molecules as distributed around the dimer center of mass; the z-axis of these cylindrically-averaged functions is the
internuclear bond axis, and the r-axis points along the bond bisector.
The right side of the TOC graphic shows the results of a classical
simulation where Na2 is described as two Lennard-Jones spheres
with the appropriate diameter and bond length (i.e., a `dumbbell' molecule).
The classical simulation gives an incorrect structure with
solvent preferentially lying in the small gap between the two atoms, where
the quantum simulation shows solvent is unlikely to go. Thus, a correct
description of solvent structure and dynamics requires that bonding
electrons be treated quantum mechanically -- classical approximations,
even highly sophisticated ones, simply cannot do the job, because they
cannot correctly account for the interaction of the solvent weith the bonding electrons
 On the experimental side of the project, graduate student Stephanie
Doan has completed preliminary studies of the steady-state and ultrafast
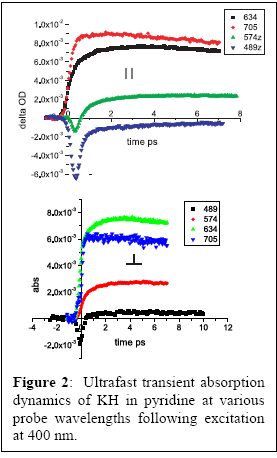
spectroscopy of alkali metal hydrides in solution. Figure 1 shows
fluorescence collection and excitation scans for KH in liquid pyridine.
Unlike gas-phase KH, which has a fluorescence quantum yield of unity for
excitation to the A-state, in solution we see an emission quantum yield that
is ≤ 1%, suggesting that the excited-state dynamics of this molecule are
entirely different in solution and that the excited molecule likely dissociates.
On the experimental side of the project, graduate student Stephanie
Doan has completed preliminary studies of the steady-state and ultrafast
spectroscopy of alkali metal hydrides in solution. Figure 1 shows
fluorescence collection and excitation scans for KH in liquid pyridine.
Unlike gas-phase KH, which has a fluorescence quantum yield of unity for
excitation to the A-state, in solution we see an emission quantum yield that
is ≤ 1%, suggesting that the excited-state dynamics of this molecule are
entirely different in solution and that the excited molecule likely dissociates.
This expectation is borne out by the pump-probe transient absorption experiments shown in Figure 2: what little stimulated emission there is decays quickly, and a large transient absorption is seen in the visible and near-IR. The dynamics suggest an excited state absorption that dynamically shifts to the blue. We are still in the process of trying to determine whether this transient species is one of the KH excited states or possibly the neutral potassium or hydride productions of dissociation, or some combination thereof.