46636-AC2
Quantum Mechanical Calculation of Hydrogen Isotope Exchange Thermodynamics and Kinetics on Petroleum Model Compounds
The first year of research into the thermodynamics and kinetics of H-D isotopic exchange on kerogen-related molecules was conducted by performing tests on the accuracy of the computational methodology employed, modeling a range of H sites within molecules, and calculating fractionation factors on alkanes of various molecular weights. This research will be used to help interpret observed H-isotope fractionation factors in ancient organic matter from sedimentary rocks. Theoretical values are needed because molecular-specific isotopic analyses have recently been made possible, but experimental values are difficult to obtain especially for specific sites within compounds. Observed fractionation trends will require knowledge of both the equilbrium fractionation factors and exchange rates as a function of temperature.
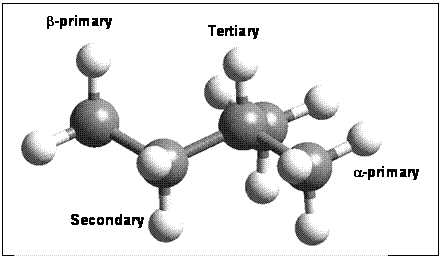
The compounds 2-methylbutane, 2,6-methyloctane, pristane and phytane were chosen because they all contain at least four distinct H sites (both a- and b-primary, secondary and tertiary – Figure 1). Thus, one can predict the relative fractionation factors and exchange rates for different site to determine whether molecular structure will affect the total H-D isotopic exchange with water. Previous work (Pedentchouk et al., 2006) has shown that branched alkanes undergo significantly more H-isotope exchange; thus we hypothesized that the fractionation factors may be greater and/or the exchange rates faster for tertiary sites than for primary sites.
Our first set of results confirms this hypothesis. For example, Table 1 shows the equilibrium fractionation factors, a-values, for our suite of compounds as a function of site within each compound. Equilibrium exchange is consistently predicted to be significantly greater at tertiary sites compared to primary sites (i.e., smaller a-values) by 50 to 70 per mil.
Table 1 – Calculated fractionation factors energies for
a suite of test compounds as calculated with quantum mechamical methods. Free energy (DG)
values are the calculated against H2O and the DG's are the differences between reactants
and products (HDO + RH ____________________________________________________________________________ Molecule/Method a-primary b-primary Secondary Tertiary 2-methylbutane CBS-Q +0.24 0.907 +0.22 0.915 +0.03 0.988 -0.10 1.043
MP2/6-31G(d,p) +0.93 0.678 +0.94 0.684 +1.10 0.643 +1.17 0.624
MP2/6-311++G(d,p) +0.95 0.683 +0.99 0.672 +1.09 0.645 +1.17 0.625
B3LYP/6-31G(d,p) +0.98 0.673 +0.96 0.680 +1.15 0.629 +1.22 0.611
B3LYP/6-311++G(d,p) +1.10 0.642 +1.07 0.650 +1.24 0.606 +1.31 0.590
2,6-methyloctane MP2/6-31G(d,p) +1.03 0.660 +0.98 0.673 +1.13 0.633 +1.24 0.606
B3LYP/6-31G(d,p) +1.07 0.649 +1.00 0.667 +1.16 0.627 +1.35 0.580
B3LYP/6-311++G(d,p) +1.12 0.635 +1.06 0.652 +1.19 0.619 +1.38 0.574
Pristane B3LYP/6-31G(d,p) +1.01 0.665 +1.02 0.663 +1.16 0.626 +1.26 0.601 B3LYP/6-311++G(d,p) +1.03 0.661 +1.07 0.648 +1.22 0.611 +1.30 0.592 Phytane B3LYP/6-31G(d,p) +1.08 0.648 +1.07 0.649 +1.19 0.619 +1.30 0.591 B3LYP/6-311++G(d,p) +0.96 0.680 +1.02 0.663 +1.11 0.639 +1.22 0.611
_____________________________________________________________________________
Figure 1 –
2-methylbutane molecule showing various types of H sites undergoing isotopic
subsitution. Frequency anlalysis with H
or D in each position gives the DG
values presented in Table 1 while energy minimizations with one H abstracted
from each site provides the energies in Table 2.
By calculating the Gibbs free
energies of H abstraction energies at each site as well, we have estimated the
activation energy barrier to H-isotope exchange. These calculations were performed with and
with H2O and the results listed in Table 2. In general, the model H
radical abstraction energies are 20 to 30 kJ/mol lower
for tertiary sites compared to primary sites.
Thus, at temperatures found in many organic-rich sediments of interest
(50 to 100°C), the rates could be different by as much as six orders of
magnitude. Obviously, this complicates
interpretations of molecular-specific H-isotope analyses because equilibrium
fractionation may be obtained at some sites and not at others. Accurate temperature estimates for the
sediments will be required in order to deconvolute this signal. The reactions modeled with H2O
present decreased the H+ abstraction energies by
approximately 100 kJ/mol which has a dramatic effect on predicted rates. Lewan (1997) has shown that water dissolved
in bitumen is capable of exchanging H-isotopes with organic compounds, so the
presence and isotopic signal of any water associated with sedimentary organic
matter must be considered when interpreting the H-isotope signal.
Table
2 – Activation barrier energies for the H abstraction from 2-methylbutane
and water reaction. The energies
are in kJ/mol. The reaction for the
activation barrier energy was 2MeBut + H2O ® 2MeBut- + H3O+.
_____________________________________________________________________ Molecule/Method a-primary b-primary Secondary Tertiary____ MP2/6-31G(d,p) +349 +276 +262 +272 MP2/6-311++G(d,p) +326 +368 +305 +298 B3LYP/6-31G(d,p) +317 +283 +228 +288 B3LYP/6-311++G(d,p) +315 +283 +227 +286 _____________________________________________________________________ References Pedentchouk N., Freeman K. H., Harris N. B. (2006)
Different response of dD values of n-alkanes, isoprenoids andkerogen during
thermal maturation Geochimica et Cosmochimica Acta, 70, 2063-2072 Lewan M.D. (1997)
Experiments on the role of water in petroleum formation. Geochimica et Cosmochimica Acta, 61, 3691-3723.