

39421-B4
Perturbations Upon Aromaticity and Antiaromaticity Due to Isotopic Substitution
Over 30 years ago a particularly useful synthesis of 1,4-dehydrobenzene (an aromatic 1,4-diradical system) was obtained from the unimolecular cycalization of 1,5-hexadiyne-2-ene.1 The resulting diradical proved viable for a plethora of uses in many areas of chemistry and biochemistry, and the so called Bergman cycalization (eq 1) became part
of the route to a variety of substances.1a
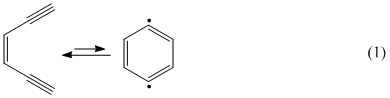
Dehydro-1,5-hexa-2-ene (1,5-hexadiyne) does not undergo an analogous cycalization. However, when treated with a strong base, under high vacuum conditions, it undergoes a bimolecular cycalization via dimerization, eq 2.
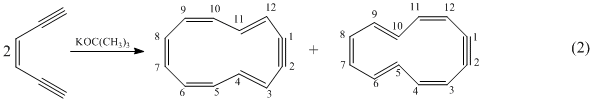
Under identical conditions (potassium tert-butoxide in THF), 1,6-heptadiyne does not dimerize; instead, it undergoes unimolecular cycalization that is somewhat analogous to that observed in the hexadiyene-2-ene system. However, the newly created ring is seven membered. Indeed, exposure of 1,6-heptadiyne, under high vacuum conditions, to a THF solution containing 18-crown-6 and potassium tert-butoxide yields a dark green solution, which persists below 220 K and yields a very weak EPR spectrum revealing coupling of a single unpaired electron to five pairs of protons. Contact of the solution with a freshly distilled potassium mirror increases the signal intensity of the EPR spectrum from this anion radical. The spectrum is that from the, previously reported,2 anion radical of cycloheptatriene.
Simply cooling an attached NMR sample tube causes the THF-d8 and a volatile hydrocarbon to cleanly distill into the NMR tube, which can subsequently be sealed from the apparatus. The now clear colorless, freshly distilled, THF-d8 solution exhibits a 1H-NMR signal that is due to cycloheptatriene.3 This spectrum reveals unprecedented resolution and long range splittings that have not been previously observed, Figure 1. Further, the same process produces cyclooctatriene from 1,7-octadiyne and cyclononatriene from 1,8-nonadiyne.
Methylene chloride in the presence of olefinic anions and strong base produces the, very reactive, ‘chlorocarbene,' which reacts with stable anions. As a result, mixtures of the cyclooctatetraene dianion and methylene chloride yield bicyclo[6.1.0]nonatriene,4 and the associated K+ ion appears to guide the attack of the CH2Cl2 anion on the triple bond of the [8]annulyne anion radical.5 Hence, we were motivated to investigate the possibility of the very transient, undergoing ring closure, heptadiyne anion capturing methylene chloride in the presence of solvated electron producing the anion radical of cyclooctatetraene.
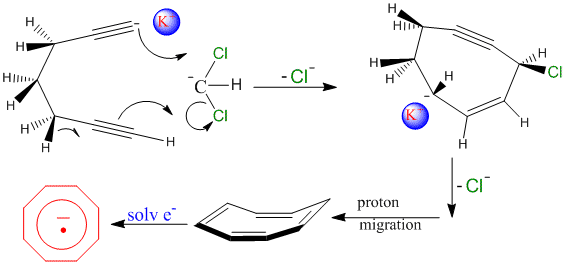
Figure 1. 400MHz 1H-NMR spectrum of cyclohetatriene in perdeuteriated benzene. This material was distilled, under vacuum, from a reaction mixture of 18-crown-6, potassium tert-butoxide and 1,6-heptadiyne in THF-D8. The small peak at d 3.2 is from the OH of tert-butanol.
Exposing pure dry HMPA to a potassium metal mirror, under high vacuum, produces a solution that exhibits the single resonance EPR spectrum of the solvated electron. Adding a mixture of heptadiyne and methylene chloride to this solution, results in the immediate disappearance of the EPR signal for the solvated electron. Repeated exposure to the potassium mirror leads to the formation of the anion radical of cyclooctatetraene, as evidenced by its, well known, nine line EPR pattern, Scheme 1.
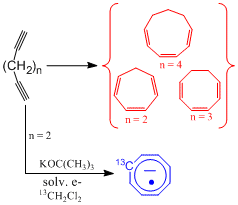
Likewise, replacing the methylene chloride with 99% carbon-13 enriched methylene chloride, results in the EPR spectrum of mono-13C-cyclooctatetraene anion radical. We feel that this cycalization is a rather unique ring closure protocol which can be used to form cycloheptatriene from heptadiyne, cyclooctatriene from octadiyne, and cyclononatriene from octadiyne. Further, the ring closure can be used to ‘pinch' and incorporate another carbon (substituted or not) atom into a ring structure that is one member larger.
References
(1) (a) Basak, A.; Mandal, S.; Bag S. S. Chem. Rev. 2003, 103, 4077-4094. (b) Bergman, R. G. Acc. Chem. Res. 1973, 6, 25.
(2) Stevenson, C. D.; Kim, Y. S. J. Am. Chem. Soc. 2000, 122, 3211.
(3) Hammons, J. H.; Hrovat, D. A.; Borden, W. T. J. Am. Chem. Soc. 1991, 113, 4500.
(4) (a) Katz T. J.; Garratt, P. J.; J. Amer. Chem. Soc., 1964, 86, 5194. (b) Vogel, E. Angew. Chem., 1962, 74, 829.
(5) Kiesewetter, M. K.; Reiter, R. C.; Stevenson, C. D. J. Am. Chem. Soc. 2005, 127, 1118-1119.