www.acsprf.org
Reports: UR449447-UR4: Investigating Proton-Coupled Electron Transfer with Radical Cations Appended with Bases
Progress in this year of the grant has focused on synthesis and characterization of radical cations PPT+, NAr2(pyOMe)+, MPT-pyr+, MPT-2-PQ+, and MPT-3-PQ+; and their reactivity with hydrogen atom donors. Such compounds combine one-electron oxidants and bases to investigate unexplored 1H+/1e reactivity and mechanisms. Such compounds may have use in hydrocarbon functionalization.
|
|
|
PPT+ | NAr2(pyOMe)+ | MPT-pyr+ |
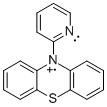
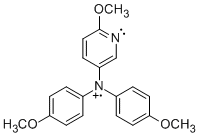
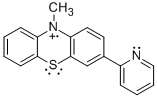
|
|
MPT-2-PQ+ | MPT-3-PQ+ |
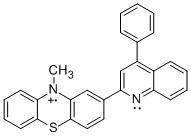
Student researchers Evan Welker and Robert Richards synthesized the precursors for radical cations PPT+ and NAr2(pyOMe)+ using procedures modified from the literature.1 We obtained MPT-2-PQ and MPT-3-PQ from Samson Jenekhe at the University of Washington. As indicated in the figure and table, cyclic voltammetry for these compounds in 0.1 M n-Bu4NPF6 in acetonitrile affords oxidation and reduction waves, indicating clean electrochemical oxidations on the timescale of the scans (although the phenylquinolinyl compounds were limited by their solubility). These potentials are similar to parallel compounds.2 The more positive potentials of PPT+/0 and NAr2(pyOMe)+/0 can be explained by the electron deficient nature of the pyridine ring.
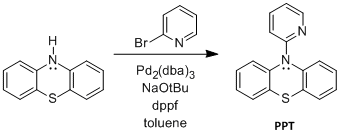
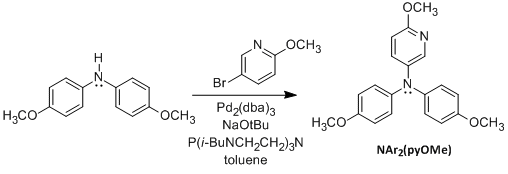
Figure 1. Syntheses of PPT and NAr2(pyOMe).
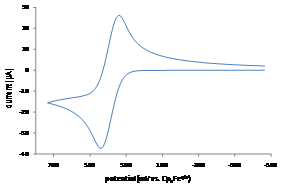
Figure 2. Cyclic voltammogram of PPT+/0.
Table. Experimental reduction potentials.
compound | E (V vs. Cp2Fe+/0) |
PPT+/0 | 0.39 |
MPT-3PQ+/0 | 0.33 |
MPT-2PQ+/0 | 0.33 |
MPT+/0 | 0.32 (ref 2) |
MPT-py+/0 | 0.32 |
NAr2(pyOMe)+/0 | 0.25 |
N(PhOMe)3+/0 | 0.16 (ref 2) |
The neutral precursors were oxidized with tris(4-bromophenyl)aminium hexafluorophosphate in acetonitrile or methylene chloride. (The hexachloroantimonate and tetrafluoroborate salts led to chlorinated and fluorinated side products, respectively, in the reactions with hydrogen atom donors, and other oxidants had unidentified side products.) Absorption maxima in the UV-vis spectra parallel those in the spectra for MPT+ and N(PhOMe)3+.3 In addition, these reactions give broad 1H NMR spectra in CD3CN. Several sources in the chemical literature state that pyridine nitrogen radical cations rapidly decompose, as the nitrogen can act as a nucleophile. All the compounds (including MPT-pyr+, a compound we continue to study) are stable for hours to days in dry acetonitrile under ambient atmosphere. All of the reactivity studies were performed with radical cations generated in situ in this manner.
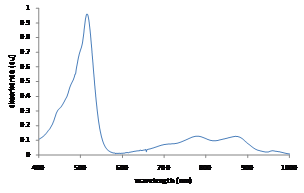
Figure 3. Visible spectrum for PPT+
Radical cations can accept an electron, and pyridine can accept a proton. Our hypothesis is that the combination of these two components will allow for the net transfer of a hydrogen atom by concerted proton-electron transfer (CPET). Hence, we explored the reactivity of these radical cations with two hydrogen atom donors, 9,10-dihydroanthracene (DHA) and 1,4-cyclohexadiene (CHD), in solution with UV-vis and 1H NMR. Initial attempts with xanthene, fluorene and indene still require further analysis.
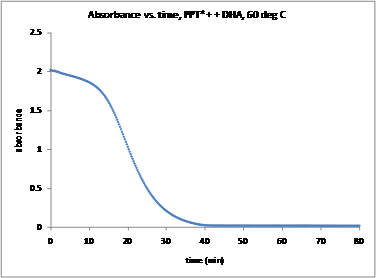
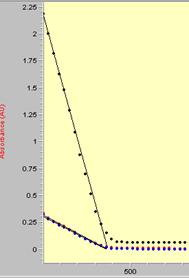
Addition of DHA or CHD to each compound in acetonitrile leads to the decay of the absorptions in the visible region of the spectrum. In reactions with DHA and CHD, peaks corresponding to anthracene and benzene, respectively, appear in 1H NMR. Kinetic studies monitoring the decay of MPT-2-PQ+, MPT-3-PQ+, and PPT+ with excess CHD and DHA did not provide the expected pseudo-first-order decays; under several conditions, sigmoidal curves or straight lines were observed. Sigmoidal curves are characteristic of an autocatalytic mechanism,4 although it is unclear if one is possible. Addition of triflic acid to the reaction mixture accelerates the decay suggesting that the protonated product (such as PPTH+) may play a role. Decay kinetics for NAr2(pyOMe)+ did not fit cleanly to a pseudo-first-order integrated rate equation.
|
|
Figure 4. Kinetics trace for the decay of PPT+ in the presence of excess DHA at 60 °C. | Figure 5. Kinetics trace for the decay of PPT+ in the presence of excess CHD at 10 °C. |
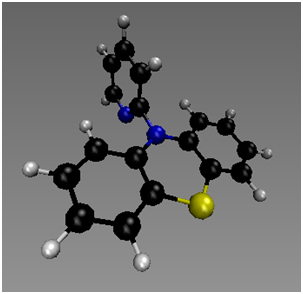
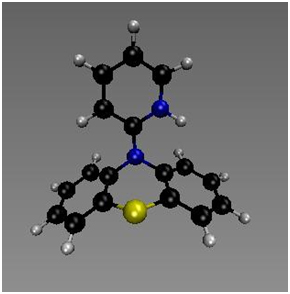
Computational studies by Jeffrey Wolbach, Samantha Cordisco and Matthew Zuchero suggest that these reactions occur by a rate-limiting CPET mechanism.5 The reactants and products for CPET mechanistic step for reaction between several radical cations and CHD and DHA have been calculated. The transition states for MPT-pyr+, MPT-2-PQ+, MPT-3-PQ+, and NAr2(pyOMe)+ + CHD and DHA have been identified and verified both with imaginary frequency and intrinsic reaction coordinate (IRC) calculations. Some intermediates for stepwise proton transfer or electron transfer have also been calculated and tentatively indicate that the concerted mechanisms occur with a much lower barrier. The transformation of PPT+ to PPTH+ requires a significant change in geometry, and hence the compound may serve as a model for conformationally gated reactions. The pyridyl ring of PPT+ is in the nodal plane of the SOMO of the system of the phenothiazium ring, and hence the concerted mechanism is separated CPET.6
|
|
Figure 6. Calculated minimum for PPT+. | Figure 7. Calculated structure for PPTH+. |
In the next year of the grant, we plan to systematically vary concentrations in kinetics runs to determine a better mechanistic model for the overall reactivity. If a CPET step is involved in the model, kinetic isotope effect studies will verify that the hydrogen atom is involved in the mechanism. Further computational studies will parallel the experiments.
(1) Urgaonkar, S.; Verkade, J.G. J. Org. Chem. 2004, 69, 9135-9142.
(2) Connelly, N. G.; Geiger, W. E. Chem. Rev. 1996, 96, 877-910.
(3) Rosokha, S. V.; Kochi, J. K. J. Am. Chem. Soc. 2007, 129, 3683-3697.
(4) King, G. A. M. Chem. Soc. Rev.1978, 7, 297-316.
(5) Geometries were calculated with B972/6-31+g(d,p) with thermal corrections with B972/6-31+g(d,p) in the gas phase. Energies for the stationary points were calculated with CAM-B3LYP/6-31++g(2d,p). Transition states were calculated with ONIOM (CAM-B3LYP/6-31++g(2d,p):B972/3-21+g), and the frequencies and IRC were calculated with ONIOM (CAM-B3LYP/6-31++g(2d,p):B972/3-21+g). The energies, transition states, frequencies and IRC were calculated with implicit solvation in CH3CN.
(6) Warren, J. J.; Tronic, T. A.; Mayer, J. M. Chem. Rev. 2010, 110, 6961-7001.